Review
The Role of the Human Papillomavirus (HPV) in Cervical Cancer: A Review about HPV-Induced
Carcinogenesis and Its Epidemiology, Diagnosis, Management and Prevention
Karl Bonello1, Renald Blundell1
doi: http://dx.doi.org/10.5195/ijms.2016.146
Volume 4, Number 1: 26-32
Received 07 09 2015:
Accepted 12 03 2016
ABSTRACT
The human papillomavirus (HPV) was the first virus known to induce carcinogenesis
and is associated with cancers of the uterine cervix, anogenital tumors and malignancies
of the head and neck. This paper reviews the structure and basic genomic characteristics
of the virus and outlines the clinical involvement of the main HPV serotypes, focusing
on the carcinogenic role of HPV-16 and 18. The mechanisms that occur in the development
of cervical neoplasia due to the oncogenic proteins E6 and E7 which interfere with
the regulation of the cell cycle through their interaction with p53 and retinoblastoma
protein are described. Epidemiological factors, diagnostic tools and the management
of the disease are also reviewed, along with the available vaccines to prevent the
viral infection. Insights on current research on involvement of oxidative stress and
micro-RNAs in cervical carcinogenesis are also explored as they may unlock new means
of diagnosis and treatment in the future.
Keywords:
Human papillomavirus 16;
Human papillomavirus 18;
Uterine Cervical Neoplasms;
Biomarkers;
Papillomavirus Vaccines.
Introduction
The human papillomaviruses (HPVs) are a group of small viruses containing deoxyribonucleic
acid (DNA) which belong to the Papillomaviridae family. They are highly ubiquitous
and properly adapted to their hosts, with the ability to effectively mask themselves
from immune responses. Over 200 types of viruses have been identified and classified
into 16 genera, and most of these affect humans. These viruses primarily target differentiating
squamous epithelium and are associated with cutaneous infections, in which almost
any part of the human skin can be affected, and mucosal infections. HPV infection
is a risk factor for malignancy of the uterine cervix by playing an integral role
in carcinogenesis through the action of its genomic products.1
HPV Structure
HPVs are spherical, non-enveloped particles with an approximate diameter of 52–55
nm and a molecular weight of about 5×106 Da. Their capsid has an icosahedral symmetry and is composed of 72 capsomeres, each
possessing five identical subunits and hence a five-fold symmetry.2 Of these capsomeres, 60 are hexavalent (exhibit six-coordination), whilst the remaining
12 are pentavalent.3
The HPV Genome
The nucleic acid core of the virion consists of supercoiled double-stranded closed
circular DNA of approximately 8,000 base pairs. The open reading frame (ORF) size
is larger than 400 bases and found on one strand exhibited in the form of arcs external
to the circular genome. All viral messenger ribonucleic acid (mRNA) transcripts are
derived from one strand.4
Both the capsid and genome morphology are similar across the Papillomaviridae family,
but differences in the genomic organization as well as size and sequences of nucleotides
and amino acids have allowed the separation of papillomaviruses into separate groups.5
The genome of all papillomaviruses consists of three unequal regions. The upstream
regulatory region (URR) is also known as the long control region (LCR) or the non-coding
region (NCR) and constitutes around 10% of the entire genome.3 The early region occupies approximately half the genome and is further divided into
two large and many smaller reading frames: E1–E2 and E4–E7, respectively.3 E6 and E7 are directly involved in the development of cervical cancer.2 The late region completes the remaining 40% of the genome and contains two genes:
L1 and L2.3
HPV Classification and Serotypes
Although HPV is commonly implicated in cases of cervical cancer, this is not the sole
clinical scenario in which the virus is involved. There is a great diversity of HPV
types that cause various lesions, which may overlap across the different types. HPV
serotypes are genetically diverse from each other and, in fact, the standard systematic
classification system states that a specific HPV type must possess a complete genome
in which the L1 nucleotide sequence differs from that in any other HPV genome by a
minimum of 10%. HPV types are denoted numerically, in the chronological order of when
they were discovered.2 There are over 200 types of HPV and these are further classified into 16 genera.1 There are further subdivisions known as HPV species, which represent serotypes that
are genetically distinct but are characterized by similar biological activity. In
addition, there are also other HPV subtypes and variants that are 2–10% and <2% dissimilar.2
Due to the fact that there is a multitude of HPV serotypes and that they have evolved
to occupy various biological niches,2 they are often divided into three groups: cutaneous, mucocutaneous, and the viruses
associated with epidermodysplasia verruciformis.1 The Beta genus encompasses the cutaneous HPVs, together with some members of the
Gamma and Mu genera. The Alpha genus is mainly characterized by the mucosal sero-types
but also some cutaneous types that cause warts.5 HPVs can also be grouped depending on the regions of the body that they infect and
the diseases they cause.2
There is yet another classification system for HPVs based on their oncogenic potential.
In this system, the viruses are divided into three groups depending on whether they
cause an infection which has a low, intermediate or high risk of resulting in a malignancy,5 as can be seen in Table 1.
Table 1.
HPV Serotypes Grouped according to Oncogenic Risk
| Risk Category |
HPV Serotype |
Relation to Cervical Cancer |
| Low |
6, 11 |
Rarely associated with cervical cancer and mostly causes genital warts |
| Intermediate |
22, 35, 39, 52, 56, 58, 59, 68 |
Present in around 25% cases and are rarer than the high-risk serotypes |
| High |
16, 18, 31, 45 |
Present in around 75% of cases, the commonest types being 16 and 18 |
Adapted from Blundell R, Camilleri G. The human papillomaviruses (HPVs) and HPV DNA
testing. In: Blundell R, ed. Infertility. Saarbrücken (Germany): Lambert Academic
Publishing; 2013. p. 57-82. Reprinted with permission from Blundell R.
Epidemiology
The anogenital type of infection is the most common sexually transmitted infection
(STI) in the United States—it is estimated that 6.2 million people acquire the infection
every year. The infection tends to be more common in adolescents and young adults.
Clinical studies have shown that the prevalence in adolescent girls is normally around
30% and can even go up to 64%. Another study explained that four years after first
sexual intercourse, over half of the young woman population were affected by cervical
HPV infection. The same study also reported that HPV can be transmitted via nonpenetrative
sexual behavior, but the probability is lower than acquiring the infection through
penetrative sex. Apart from sexual behavior and history, studies in the United States
reported that an age of less than 25 years is also a risk factor for the infection
and the prevalence tends to decrease after this age, except for one cohort study in
Costa Rica, which found that the prevalence rises again after 40. Furthermore, men
and women seem to have similar rates of HPV infection.6 Figure 1 illustrates the prevalence of HPV infection according to age.
Figure 1.
Prevalence of High-Risk HPV Infection among Females Undergoing Cervical Screening
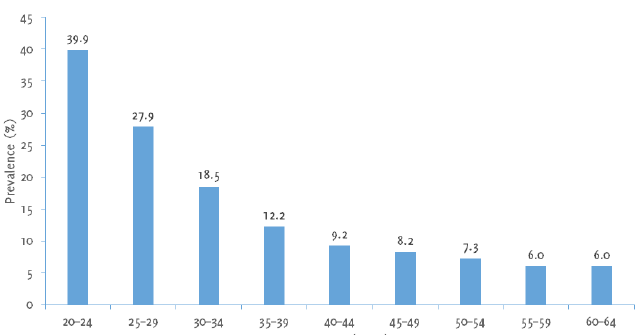
Compiled using information from Kitchener H, Almonte M, Wheeler P, Desai M, Gilham
C, Bailey A et al. HPV testing in routine cervical screening: cross sectional data
from the ARTISTIC trial. Br J Cancer. 2006;95(1):56-61.
The detection of high-risk HPV in virgins, neonates and children provides evidence
that the sexual route is not the sole means of HPV transmission. Both low- and high-risk
HPV serotypes can be transmitted via non-sexual routes such as physical contact, shared
bathing and infected fomites. It appears that the transmission of high-risk HPV from
mother to infant occurs during parturition as the neonate descends through the infected
birth canal. This is more likely to occur in women having markedly higher amounts
of HPV DNA, such as HPV-16, than in those having less DNA. Furthermore, a case of
two mothers with both HPV-16 and 18 infections gave birth to neonates with similar
co-infection.7
The state of pregnancy seems to increase the likelihood of developing genital HPV
infections. This was demonstrated by a study which showed that 52.5% of patients tested
positive for HPV DNA during the third trimester of pregnancy, compared to 17.5% after
parturition. Physiological explanations for this finding may be that the hormone profile
during pregnancy increases the transcription of HPV genes due to interactions between
glucocorticoids and their response elements present in the non-coding region of HPV-16.
Additionally, pregnancy is a state of immunosuppression. HPV infections can also be
transmitted during pregnancy itself across the placenta and through amniotic fluid.
In fact, a study revealed the presence of HPV DNA in 75% of amniotic fluids taken
from women who were positive for cervical HPV DNA.7
Interestingly, the first evidence that suggested an association between HPV and cancer
emerged from research in the 1970s on skin cancer in patients suffering from epidermodysplasia
verruciformis,4 which is a rare autosomal recessive disorder associated with the mutation of one
of the two genes: EVER1 and EVER2 located on chromosome 17.1 This disorder is characterized by flat warts or pityriasis-like lesions that are
caused by infections with HPV types 5, 8 or 17. These warts resulted in skin cancers
in 20% of patients, mostly in areas exposed to the sun.4 It appears that the mutated genes code for transmembrane proteins that function in
zinc homeostasis, and their loss results in the transcription of the E6 and E7 genes,1 thereby facilitating carcinogenesis.
Cervical cancer is the second most common malignancy in women around the world and
affects an average of 35 per 100,000 women.1 The interest in the association between HPV and cancer was greatly magnified when
HPV types 16 and 18 were discovered in cervical cancers and preneoplastic dysplasia,
lesions that can predispose one to malignancy of the uterine cervix.4 It has been found that HPV DNA features in over 99% of cervical cancer cases; however,
the most common high-risk serotype varies across countries, ethnicities and socioeconomic
statuses. In a study of more than 30,000 cervical cancers, the International Agency
for Research on Cancer (IARC) demonstrated that amongst the most common HPV serotypes
responsible for causing cervical malignancy (16, 18, 58, 33, 45, 31, 52, 35, 59, 39,
51, 56), HPV 16 causes over 50% whilst HPV 16 and 18 cause more than 70% of cases
worldwide. HPV serotypes 18 and 45 are implicated in the more aggressive cervical
adenocarcinomas.1
It has been found that sustained infection with high-risk HPV serotypes is the most
fundamental risk factor in the development of precursor lesions for cervical cancer.
Persistence is normally defined as the detection of the same high-risk HPV types at
>2 visits 4–6 months apart.6 In fact, studies have shown that such persistent infections can increase the likelihood
of developing high-grade precursors of cervical malignancy by more than 10 times.6
Pathophysiology
The Cervical Canal and Associated Malignancies
The cervix represents the anatomical transition between the vagina and the uterus.
It consists of a canal with two openings: the superior internal os which leads into
the uterus and the inferior external os that opens into the vaginal cavity. The histology
of the cervical canal is characterized by simple columnar secretory epithelium, as
opposed to the vaginal cavity, which is lined by stratified non-keratinizing squamous
epithelium. The epithelia lining the endocervix and exocervix meet at a point known
as the transition zone, or squamocolumnar junction which corresponds to the region
of the cervix at the external os.8
The squamocolumnar junction is a highly important cytological landmark because it
is the site with greatest susceptibility to infection by HPV and where over 90% of
malignancies from the lower genital tract arise. HPV is known to cause cervical dysplasia
and cervical intraepithelial neoplasia (CIN), which are likely to progress to cervical
cancer as a result of sustained infection by high-risk HPV.9
As the transition zone involves two epithelial cell types—the glandular and squamous
cells—two types of cancers can arise in the cervix. The uncontrolled proliferation
of the glandular cells of the endocervix produces an adenocarcinoma (10–20% of cases,
but incidence seems to be on the rise in recent years), whereas a malignancy of the
squamous cells results in squamous cell carcinoma. The latter is far more common (80–90%
of cases) and is often asymptomatic at its initial stages, but can present with coital
and pelvic pain as well as aberrant vaginal bleeding and discharge at later stages.
Life Cycle of the HPV
The process of cervical carcinogenesis is intimately linked to the events occurring
in the life cycle of the virus, as shown in Figure 2. In a stratified squamous epithelium, the cells forming the basal layer behave as
stem cells and therefore have the function to undergo cell division in view of replenishing
the cells shed from the surface layer. When the basal cell divides by mitosis, two
daughter cells are produced: one ascends and transforms into a terminally differentiated
cell, whilst the remaining cell resides in the basal layer to maintain the pool of
dividing cells. The first targets of the virus are in fact the basal cells that are
accessed through microwounds. HPV virions enter the cells by interacting with specific
receptors like the alpha-6 integrin that binds HPV-16. Viral DNA replication commences
in the basal layers, producing 50–100 copies of the genome in each cell. This is accompanied
by the expression of the E1 and E2 proteins, which are necessary for the replication
process and for the segregation of newly synthesized DNA, thus ensuring that infected
stem cells remain in the lesion for a prolonged timeframe. The virus mainly utilizes
host machinery to carry out DNA replication, except for the E1 helicase. The products
of early genes such as E5–E7 are thought to create an optimal environment for replication
to occur, for example, by stimulating DNA replication in the host cell and preventing
apoptosis.9
Figure 2.
Life-cycle of the Human Papillomavirus
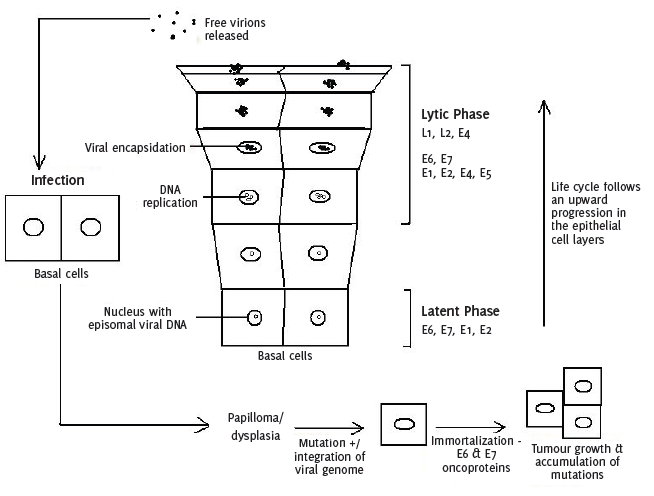
Compiled using information from Narisawa-Saito M, Kiyono T. Basic mechanisms of high-risk
human papillomavirus-induced carcinogenesis: roles of E6 and E7 proteins. Cancer Sci.
2007 Oct;98(10):1505-11.
As the infected basal cells migrate upwards and differentiate, the viral late genes
L1 and L2 are transcribed, thereby inducing the vegetative stage of the life cycle
characterized by drastic amplification of the genome.9 It appears that the control of the expression of the late genes depends on the state
of differentiation of the host cell.4 Once the cell reaches the outermost layer of the epithelium, the newly synthesized
viral DNA is encapsidated to form new virions, which are released, and the life cycle
is then repeated. As HPVs do not induce complete lysis in host cells, new virions
are deposited in squames that are continuously shed.10 It is interesting to note that the virus is well secluded from the host immune system,
as the immunogenic virions are only assembled in the outer layers of the epithelium.
Furthermore, the viral proteins E6 and E7 also function in keeping the infection asymptomatic
by inactivating interferon regulatory factor.9
Squamous epithelial cells that have been infected by HPV undergo koilocytosis to form
cells called koilocytes. These cells have an enlarged, darkened and irregularly outlined
nucleus surrounded by an area of clear space called a perinuclear halo and appear
vacuolated. This transformation indicates mild cellular dysplasia and represents a
highly replicative viral state. When the extent of dysplasia is moderate or severe,
cells appear small and multiply even on the outermost layer of the epithelium, making
the lesion possibly carcinogenic if severe.11
The Role of HPV in Cervical Carcinogenesis
Although HPV is the major risk factor for cervical cancer, many researchers speculate
that the actual integration of viral DNA in the host cell is not a common event and,
most of the time, HPV infection is resolved relatively quickly by the immune system.
Even though the viral DNA can cause rapid neoplastic transformation of the infected
cells once integrated, the presence of HPV DNA in the cell alone is not sufficient
to cause cancer because other genetic and epigenetic events are probably required.9
The two major oncogenic protein products of the HPV virus are E6 and E7, which function
by altering the regulation of the cell cycle and the control of apoptosis. The integration
of viral DNA disturbs the activity of the E2 protein. This protein is known to repress
the transcription of E6 and E7 and, therefore, its disruption leads to dysregulated
expression of the oncoproteins. Together, these proteins are capable of causing immortalization
of cells, which maintain their mitotic ability to produce clones which also possess
the immortalized phenotype and fail to undergo terminal differentiation.4
The E7 Oncoprotein
The major characteristic of E7 that enables it to enhance neo-plastic change is its
interaction and subsequent inactivation of pRb. The state of phosphorylation of pRb
changes according to the phase of the cell cycle, being dephosphorylated in G0-and
G1-phases, phosphorylated throughout the S-phase and remains thus until late in the
M-phase when it assumes the hypophosphorylated form once again via a specific phosphatase.4
When dephosphorylated, pRb and associated proteins inhibit transcription factors like
E2F by binding to them, thereby repressing the expression of genes whose products
stimulate DNA synthesis and enhancing the progression of the cell cycle. Conversely,
when pRb is phosphorylated by G1 CDKs, it is unable to bind to E2F and its inhibitory
effect is thus lifted, allowing the cell to proceed to the S-phase. It is therefore
a regulator of the G1/S checkpoint. Detection of damaged DNA results in the activation
of p53, which in turn activates p21, a CDK-inhibitor. This binds to and inhibits cyclinE-CDK2
and, hence, pRb cannot be phosphorylated, In other words, it can inactivate E2F and
block the G1/S transition.12
The E7 protein can bind to the hypophosphorylated form of pRb. This interferes with
the complex formed between pRb and E2F, resulting in the premature progression of
the cell into S-phase, thereby favoring DNA synthesis and, consequently, cell division.
E7 is also involved in the enhancement of pRb cleavage from its C-terminal by calpain,
which is a cysteine protease activated by calcium. This is in fact a step that must
precede the E7-induced proteasomal cleavage of pRb, which involves the 26S proteasome.9 Interestingly, the actual production of E7 and its effects on targets like pRb are
necessary for the replication and completion of the full life cycle of HPV.10
The E6 Oncoprotein
The E6 protein primarily exerts its neoplastic effect on HPV-infected cells by promoting
the ubiquitin-dependent proteosomal degradation of p53,9 which is a tumor suppressor gene product that protects the cell against the accumulation
of harmful mutations that can lead to cancer development. Such mutations can be due
to DNA damage by physical and chemical mutagens, as well as errors during DNA replication.
When abnormal DNA is detected and p53 is activated, the cell cycle is paused, allowing
DNA repair to occur before the cell divides. In certain circumstances, such as when
DNA cannot be effectively repaired, apoptosis can be induced for programmed cell death.10
The concentration of p53 in cells containing E6 such as cervical cancer cells is around
2–3 times smaller compared to uninfected cells. Its half-life is also markedly reduced.
As a consequence, the normal response to DNA damage mediated by p53 is not carried
out. DNA mutations reside in the genome uncorrected and are passed on from one cellular
generation to another, resulting in their accumulation over time and hence causing
genomic instability.4 Apart from the lack of checkpoint surveillance for DNA damage in cancer cells, they
also have an intrinsic tendency to favor mutagenesis.10
The binding of E6 to p53 is not direct and is mediated by E6-associated protein (E6AP),
which is an E3 ubiquitin protein ligase. This is part of a class of proteins homologous
to the E6-AP carboxyl terminus (HECT) E3 ligases that function in the recognition
of substrates by ubiquinylation machinery targeted for proteosomal degradation. Interestingly,
the presence of E6 increases the turnover of E6AP, probably as a result of its enhanced
enzymatic activity in the HPV-infected cellular environment.4
Areas of Current Research
Current research is focusing on the role played by oxidative stress in HPV-mediated
carcinogenesis. Reactive oxygen species induce DNA damage and modulate the viral life
cycle. Studies have proposed a link between the oxidative status of infected cells
and the persistence or progression of the lesion. Oxidative stress also has pro-survival
and anti-apoptotic effects on infected cells and contributes to the expression of
the E6 and E7 genes. This appears to be a promising area of study, especially to determine
the effects of oxidative stress on chemo-therapy response and resistance and its potential
as a marker for diagnostic purposes.13
Micro-RNAs (miRNAs) appear to have a role in cervical carcino-genesis. They are short
strands of non-coding RNA that normally have short half-life and alter gene expression
post-transcriptionally. Their deregulation is associated with the onset, progression
and metastasis of human tumors, including cervical cancer which exhibits increased
and decreased levels of oncogenic or tumor suppressive miRNAs, respectively. The E6
and E7 proteins are able to modulate miRNAs. It is therefore useful to determine how
they can be used as prognostic biomarkers by comparing miRNA profiles of normal and
transformed cells. In addition, future studies can possibly unlock another form of
cervical cancer treatment involving RNA modification therapy.14
Diagnosis
Cytology and Histology
The introduction of screening programs over the past decades has succeeded in reducing
the incidences and mortalities of cervical cancer.15 The most common and convenient technique used to assess the cervix for dysplastic
cellular changes is the Papanicolau smear. In this procedure, the vaginal orifice
is held open by a speculum, and a sample of cells is taken from the squamocolumnar
junction by inserting and rotating a spatula through the external os. The smear is
fixed onto a slide, stained appropriately and observed under the microscope to observe
the morphology of the epithelial cells. The use of a colposcope allows the direct
observation of the cervix with a magnified epithelium and the possibility of taking
a biopsy.1 It has been estimated that 40–50% of cervical cancers are diagnosed in women who
take routine cervical cytology screening16 and that four out of five women diagnosed with cancer had not taken the test in the
previous five years.1
Liquid-based cytology has permitted the optimization of the quality and consistency
of samples by creating uniform cell layers and decreasing the number of poor samples.
The presence of koilocytes is indicative of productive HPV infection, whilst persistent
infection manifests itself as highly severe changes in the nucleus, mitotic figures
and aggregates of pyknotic cells. Different classification methods are used for these
observations (see Table 2), but the Bethesda classification is often the preferred one and presents two categories:
(1) low grade squamous epithelial neoplasia (LSIL), which encompasses samples with
cells having irregular, larger and well-defined nuclei and (2) high grade squamous
epithelial neoplasia (HSIL) for samples characterized by poorly differentiated and
immature, small cells having distinct cytoplasmic borders and organized in sheets
and syncytial groups.1
Table 2.
Classification of Cervical Intraepithelial Lesions
| Papanicolau Classification |
Dysplasia Classification |
Bethesda Classification |
Histology Classification |
| I |
Negative squamous atypia |
NILM (Negative for intraepithelial lesion or malignancy) |
Negative |
| II |
Squamous atypia |
ASCUS (Atypical squamous cell of unknown significance), ASC-H (Atypical squamous cells
– cannot exclude HSIL)
|
Squamous atypia |
|
Mild |
LSIL (Low grade squamous intra-epithelial lesions) |
CIN1 (Abnormal cells in 1:3 of layers; very unlikely to progress) |
| III |
Moderate |
HSIL (High grade squamous intra-epithelial lesions) |
CIN2 (Abnormal cells including mitotic figures in 2:3 of layers with loss of stratification
and differentiation) CIN3 (Abnormal cells in all layers; can progress to invasive
cancer if untreated)
|
| IV |
Severe CIS (Carcinoma in situ)
|
HSIL |
CIN3 (Abnormal cells in all layers; can progress to invasive cancer if untreated) |
| V |
Carcinoma |
Carcinoma |
Carcinoma |
Compiled using information from Cubie HA. Diseases associated with human papillomavirus
infection. Virology. 2013 Oct;445(1-2):21-34.
Histological assessment of samples provides more specific information about the HPV
infection by revealing features such as basal hypertrophy, excess surface keratinization
and general disruption of the epithelium. Cervical intraneoplasia lesions are graded
according to the fraction of epithelium that exhibits abnormalities.1
Biomarkers
There are various biomarkers that can be used to identify the presence and extent
of HPV infection and progression of cervical malignancy. They can be divided into
three groups. The first group comprises HPV DNA, RNA and proteins, which prove to
be highly sensitive and specific for the diagnosis of CIN2 or worse lesions in women
of 30 years or older and in detecting adenocarcinoma. The second group involves cellular
biomarkers, which are based on the fact that the E6 and E7 proteins alter several
pathways that control the cell cycle. For example, as the inactivation of pRb leads
to an increase in the CDK inhibitor p16, the overexpression of this molecule can be
detected by immunostaining or enzyme-linked immunosorbent assay (ELISA) and hence
serves as a marker of cellular transformation. The last group includes epigenetic
biomarkers which reflect the methylation of DNA and hence whether DNA is active or
silenced. The state of methylation of the L1 gene appears to be associated with the
diagnosis of CIN2.17 Table 3 summarizes the different types of biomarkers used in the detection of cervical cancer
lesions.
Table 3.
Cervical Cancer Biomarkers
| Type of Biomarker |
Test/Technique |
Remarks |
| HPV DNA testing |
Hybrid Capture 2 (Qiagen) |
Detects 13 high-risk HPVs |
| Cervista HPV HR (Hologic) |
Detects 14 high-risk HPVs |
| Cervista HPV 16/18 (Hologic) |
Specifically identifies HPV 16 and 18 |
| Cobas 4800 HPV (Roche Diagnostics) |
Targets 14 high-risk HPVs |
| HPV RNA testing |
APITMA (GenProbe) and OncoTect (IncellDX) |
Based on reverse transcriptase and PCR technique and detect E6/E7 mRNA from 14 and
13 high-risk HPA sero-types, respectively
|
| PreTect HPV-Proofer (Nor-chip) and NucliSENS EasyQ (Biomerieux) |
Rely on nucleic acid sequence-based amplification (NASBA) and are able to detect E6/E7
transcripts from HPV 16, 18, 31, 33, 45
|
| HPV protein testing |
OncoE6 (Arbor Vita Corporation) |
Detects E6 protein encoded by HPV 16, 18, 45 |
| Cytoactiv Assay (Cytoimmune Diagnostics) |
Measures loss of expression of L1 which has been identified as a potential marker
progressive lesions
|
| Cellular biomarkers |
P16/K1-67 Immunocyto-chemistry Assay |
p16 is a CDK-I whilst K1-67 is a proliferation antigen expressed in the G2 and M phases
of the cell cycle. They are co-expressed in dysplastic lesions and constitute a highly
sensitive and specific test for CIN2 or worse lesions
|
| ProExCTM Assay (Becton-Dickinson) |
Recognizes minichromosome maintenance protein 2 and topoisomerase II α, which are
expressed in cells with abnormal S-phases such as HPV-infected cells with increased
E6/E7 synthesis.
|
| Fluorescence In Situ Hybridization (FISH) and Multiplex Ligation-Dependent Probe Amplification
(MLPA)
|
Can be used to detect gain of chromosomes 3q and 5p which carry the TERC and TERT
genes
|
| Epigenetic biomarkers |
Differential Methylation Hybridization (DMH) |
Allows identification of SOX1, NKX6-1, PAX1, WX1 and LMXIA genes that are often methylated
in cervical cancer and precancerous lesions
|
| Restriction Landmark Genomic Scanning (RLGS) |
Detection of methylated NOL4 and LHFPL4 genes |
| Demethylating agent + expression microarray |
Identifies methylation of SPARC and TFP2 genes |
Compiled using information from Tornesello ML, Buonaguro L, Giorgi-Rossi P, Buonaguro
FM. Viral and cellular biomarkers in the diagnosis of cervical intraepithelial neoplasia
and cancer. Biomed Res Int. 2013;2013:519619.
Management
The prognosis of cervical cancer and the appropriate treatment modality depend on
the stage of the cancer, which represents the extent of invasion and spread of the
tumor. Although cervical cancer is staged clinically according to International Federation
of Obstetrics and Gynecology guidelines, it is classified in three sub-groups for
treatment purposes.18 The earliest stages (IA1 – IIA1) represent tumors of the upper one-third of the vagina measuring <4 cm, which are
treated surgically by conization with sufficient excision margins for IA1 and conization and simple/radical hysterectomy with pelvic lymphadenectomy for IIA2. Small volume macroscopic disease (IIA1 - IB1) is treated with radical hysterectomy, and radical trachelectomy with lymphadenectomy
is used to preserve fertility.15 Intermediate stages (IB2–IVA) require primary chemoradiation,18 while advanced stages (IVB) and patients with persistent incurable disease receive
systemic chemotherapy, including cisplatin, carboplatin, paclitaxel, topotecan and
gemcitabine.19
Current research is focusing on new molecular targets for cervical cancer treatment,
such as the use of epidermal growth factor receptor (EGFR) antagonists like panitumumab
and multitargeted tyrosine kinase inhibitors including imatinib and subitinib.18,20 In addition, epigenetic therapy appears to be a promising and effective form of treatment
for cervical cancer.21 Investigations on the effects of adding the antiangiogenesis agent bevacizumab to
chemotherapy used in advanced or recurrent disease are also being carried out.22
Prevention
The HPV vaccine is an important means of preventing cervical cancer, especially if
combined with a healthy sexual lifestyle and appropriate use of contraceptives. There
has been increasing consideration that vaccinating men against HPV is a wise decision
as it serves a two-fold purpose. As HPV serotypes 16 and 18 are implicated in 70%
of anal cancers and precancerous lesions of the penis, the vaccine can reduce the
incidence of these malignancies especially in homosexual males who are at increased
risk of anal dysplasia. This is also relevant for the heterosexual population which
practices anal intercourse, and it has been found that 35.9% of women and 42.3% of
men between 18–44 years have anal intercourse with partners of the opposite sex.23 Furthermore, vaccinating men is also an indirect means of preventing HPV-induced
cervical cancer in women, as it is a sexually transmitted disease.24 Unfortunately, people in low-income countries are less likely to be vaccinated against
HPV due to its relatively high cost. In addition, most women in these countries do
not have access to routine screening, with uptake of Pap smear estimated to be 5%
within the past five years.25 This results in an overall higher mortality rate from cervical cancer in developing
countries.
HPV Vaccines
There are currently two available vaccines against HPV: Gardasil® by Merck Frost and Cervarix® by GlaxoSmithKline. Both vaccines contain virus-like particles (VLPs) that are synthesized
using recombinant DNA technology and consist of the L1 protein component of the viral
capsids.25 Such VLPs cause great response by the immune system and result in antibody titers
which are much higher than those induced by the natural infection.16
Quadrivalent Vaccine
Gardasil® is said to be a quadrivalent vaccine because it targets four HPV serotypes, namely
6, 11, 16 and 18. The L1 capsid proteins are synthesized in yeast cells (Saccharomyces cerevisae) and combined with an aluminum adjuvant. It is recommended for both sexes of ages
9–26 years and is administered in three doses at 0, 2 and 6 months. This vaccine can
grant protection against ongoing HPV infection, precancer lesions of the cervix, vulva
and vagina, as well as genital warts due to serotypes 11, 16 and 18 in females of
ages 16–26 years without previous infection.26
Bivalent Vaccine
On the other hand, Cervarix® is a bivalent vaccine which grants protection against two serotypes—HPV 16 and 18.
The VLPs in this preparation are produced in insect cells using baculovirus with an
adjuvant consisting of ASO4 and monophosphoryl lipid A from bacterial cell walls.16,26 The vaccine is indicated for girls/women aged 10–25 years and is administered in
three doses at 0, 1 and 6 months. Randomized, controlled and double-blind studies
with women aged 15–25 years demonstrated that the vaccine is efficient in preventing
precursor lesions of cervical cancer due to HPV 16 and 18 in women having no previous
infection.26
When observing the variation of antibody titer with time after the Gardasil® vaccine is administered, it is interesting to note that the variation in titer values
after the Cervarix® vaccine follows the same profile, except for two differences. First, 24 months after
vaccination with Gardasil®, 96% of participants tested positive for HPV types 6, 11 and 16 antibodies and 68%
for type 18. Seropositivity reaches 100% for both HPV-16 and HPV-18 antibodies at
51–53 months after vaccination. Second, the plateau level reached with Gardasil® is similar to the titers that are naturally induced by types 6 and 18 and greater
for types 11 and 16, whereas the plateau level with Cervarix® is much higher than that caused by the natural infection.16
Contraindications and Side Effects
The vaccine is contraindicated in people with allergies to any of its components,
and women should not be vaccinated during pregnancy even though it does not appear
to increase the incidence of miscarriage.26,27 Furthermore, people with immediate hypersensitivity towards yeast should not receive
the Gardasil vaccine. The vaccine is not contraindicated for immunosuppressed individuals
or those with previous HPV infection, although immunogenicity is not guaranteed for
the former class. Other vaccines may be administered before, during and after the
course of HPV vaccination.26 Side effects which have been reported for the quadrivalent vaccine are: (1) pain,
swelling and redness at the injection site and headache in more than 10% of receivers;
(2) bruising, itching, fever and nausea in more than 0.1%; (3) urticaria in less than
0.1%; and (4) bronchospasm in less than 0.01%.28
Conclusion
HPV infection plays an integral role in the development of cervical neoplasia. However,
modern screening procedures coupled with the promotion of safe sexual practices and
HPV vaccination can reduce the burden of this disease.
Acknowledgments
None.
Conflict of Interest Statement & Funding
The author has no funding, financial relationships or conflicts of interest to disclose.
Author Contributions
Conceptualization, Data collection, Data analysis and interpretation, Writing, Critical
revision of the manuscript, Approval of the final version: KB, RB.
References
1. Cubie HA. Diseases associated with human papillomavirus infection. Virology. 2013 Oct;445(1–2):21–34.
2. Cubie HA, Cuschieri KS, Tong CYW. Papillomaviruses and polyomaviruses. In: Greenwood D, Barer M, Slack R, Irving W, eds. Medical microbiology. 18th ed. United Kingdom: Churchill Livingstone Elsevier; 2012.
3. IARC Working Group on the Evaluation of Carcinogenic Risks to Humans. Human papillomaviruses. United Kingdom: World Health Organization, International Agency for Research on Cancer; 1995.
4. Howley PM. Warts, cancer and ubiquitylation: lessons from the papillomaviruses. Trans Am Clin Climatol Assoc. 2006;117:113–26; discussion 126–7.
5. Blundell R, Camilleri G. The human papillomaviruses (HPVs) and HPV DNA testing. In: Blundell R, ed. Infertility. Saarbrücken (Germany): Lambert Academic Publishing; 2013. p. 57–82.
6. Dunne EF, Markowitz LE. Genital human papillomavirus infection. Clin Infect Dis. 2006 Sep 1;43(5):624–9.
7. Cason J, Rice P, Best JM. Transmission of cervical cancer-associated human papilloma viruses from mother to
child. Intervirology. 1998;41(4–5):213–8.
8. Ferenczy A, Wright TC. Anatomy and histology of the cervix. In: Blaustein A, Kurman RJ, ed. Blaustein's pathology of the female genital tract. USA: Springer; 2002. p. 207–24.
9. Narisawa-Saito M, Kiyono T. Basic mechanisms of high-risk human papillomavirus-induced carcinogenesis: roles of
E6 and E7 proteins. Cancer Sci. 2007 Oct;98(10):1505–11.
10. Munger K, Basile JR, Duensing S, Eichten A, Gonzalez SL, Grace M, et al. Biological activities and molecular targets of the human papillomavirus E7 oncoprotein. Oncogene. 2001 Nov 26;20(54):7888–98.
11. Blundell R, Camilleri G. The mechanisms of HPV-induced carcinogenesis and the HPV vaccine. In: Blundell R, ed. Infertility. Saarbrücken (Germany): Lambert Academic Publishing; 2013. p. 83–100.
12. Shackelford RE, Kaufmann WK, Paules RS. Cell cycle control, checkpoint mechanisms, and genotoxic stress. Environ Health Perspect. 1999 Feb;107 Suppl 1:5–24.
13. De Marco F. Oxidative stress and HPV carcinogenesis. Viruses. 2013 Feb 12;5(2):708–31.
14. Gomez-Gomez Y, Organista-Nava J, Gariglio P. Deregulation of the miRNAs expression in cervical cancer: human papillomavirus implications. Biomed Res Int. 2013;2013:407052.
15. Martin-Hirsch PL, Wood NJ. Cervical cancer. BMJ Clin Evid. 2011 Jul 27;2011:0818.
16. Dawar M, Deeks S, Dobson S. Human papillomavirus vaccines launch a new era in cervical cancer prevention. CMAJ. 2007 Aug 28;177(5):456–61.
17. Tornesello ML, Buonaguro L, Giorgi-Rossi P, Buonaguro FM. Viral and cellular biomarkers in the diagnosis of cervical intraepithelial neoplasia
and cancer. Biomed Res Int. 2013;2013:519619.
18. Duenas-Gonzalez A, Serrano-Olvera A, Cetina L, Coronel J. New molecular targets against cervical cancer. Int J Womens Health. 2014 Dec 5;6:1023–31.
19. Scatchard K, Forrest JL, Flubacher M, Cornes P, Williams C. Chemotherapy for metastatic and recurrent cervical cancer. Cochrane Database Syst Rev. 2012 Oct 17;10:CD006469.
20. Candelaria M, Arias-Bonfill D, Chavez-Blanco A, Chanona J, Cantu D, Perez C, et al. Lack in efficacy for imatinib mesylate as second-line treatment of recurrent or metastatic
cervical cancer expressing platelet-derived growth factor receptor alpha. Int J Gynecol Cancer. 2009 Dec;19(9):1632–7.
21. Duenas-Gonzalez A, Lizano M, Candelaria M, Cetina L, Arce C, Cervera E. Epigenetics of cervical cancer. An overview and therapeutic perspectives. Mol Cancer. 2005 Oct 25;4:38.
22. Tewari KS, Sill MW, Long HJ 3rd, Penson RT, Huang H, Ramondetta LM, et al. Improved survival with bevacizumab in advanced cervical cancer. N Engl J Med. 2014 Feb 20;370(8):734–43.
23. Fau CC, Chandra AF, Febo-Vazquez I. Sexual behavior, sexual attraction, and sexual orientation among adults aged 18–44
in the United States: data from the 2011–2013 National Survey of Family Growth. National Health Statistics Reports JID - 1014795190115.
24. Geipert N. Vaccinating men for HPV: new strategy for preventing cervical cancer in women? J Natl Cancer Inst. 2005 May 4;97(9):630–1.
25. Katz IT, Wright AA. Preventing cervical cancer in the developing world. N Engl J Med. 2006 Mar 16;354(11):1110.
26. Mello CF. Vaccination against human papillomavirus. Einstein (Sao Paulo). 2013 Dec;11(4):547–9.
27. Schiller JT, Castellsague X, Garland SM. A review of clinical trials of human papillomavirus prophylactic vaccines. Vaccine. 2012 Nov 20;30 Suppl 5:F123–38.
28. Kitchener H, Almonte M, Wheeler P, Desai M, Gilham C, Bailey A et al. HPV testing in routine cervical screening: cross sectional data from the ARTISTIC
trial. Br J Cancer. 2006;95(1):56–61.
Karl Bonello, 1 Faculty of Medicine & Surgery, University of Malta, Msida, Malta.
Renald Blundell, 1 Faculty of Medicine & Surgery, University of Malta, Msida, Malta.
About the Author: Karl Bonello is currently a fifth-year medical student of a five-year program at
the University of Malta Medical School, Msida, Malta. He is also a recipient of a
Dean’s Award during his second year of study and an Academic Award from the University
of Malta Student Council in his fourth year. He ranked first place in his year group
over the first four years of the program.
Correspondence Karl Bonello. Address: Faculty of Medicine & Surgery, University of Malta, Msida
MSD 2080, Malta. Email: kbonello94@gmail.com
Cite as: Bonello K, Blundell R. The role of the human papillomavirus (HPV) in cervical cancer: a review about HPV-induced carcinogenesis and its epidemiology, diagnosis, management and prevention. Int J Med Students. 2016 Jan-Apr;4(1):26-32.
Copyright © 2016 Karl Bonello, Renald Blundell
International Journal of Medical Students, VOLUME 4, NUMBER 1, April 2016