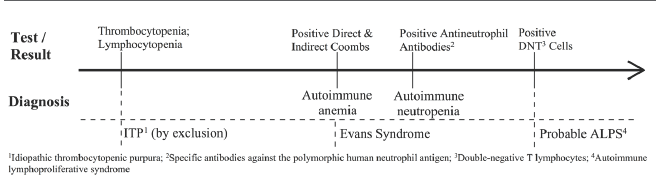
Pertinent Positive Laboratory Test Results Showing the Progression of Likely Diagnoses in a Patient with Probable ALPS
Yazan S. Mousa1
doi: http://dx.doi.org/10.5195/ijms.2016.165
Volume 4, Number 3: 123-126
Received 11 01 2016: Accepted 14 11 2016
ABSTRACT
Background:Autoimmune lymphoproliferative syndrome (ALPS) is a rare disorder of the blood, estimated at around 500 cases worldwide. It is characterized by a dysregulation of T-cells in the immune system, and is caused by a defect in the process that mediates leukocyte apoptosis. This may result in an increased risk of lymphoma and autoimmune diseases.
Case:The author reports a case of an 11-year-old male who had been followed up since three years of age for recurrent cytopenias, occurring with intermittent breakouts of purpuric rash, nosebleeds, and prolonged infections.
Conclusion:A probable diagnosis was made through criteria based on the First International ALPS workshop of 2009. This includes the presence of circulating double-negative T cells, considered the laboratory marker unique for ALPS. The mainstay of treatment was prednisone, given at doses varying in proportion to the severity of immunocytopenia. osis.
Keywords: Autoimmune Lymphoproliferative Syndrome; Autoimmunity; Pancytopenia.
Autoimmune lymphoproliferative syndrome (ALPS) is an abnormality of lymphocyte homeostasis, caused by a disruption in the apoptosis pathway mediated by the FAS gene. Clinical sequelae are most commonly defined by a lymphoproliferative pattern in early disease onset age groups, and an autoimmune pattern in later disease onset.1
The exact incidence and prevalence of ALPS are unknown. It is estimated that the number exceeds 500 worldwide, but due to difficulty in diagnosis and features that overlap with other diseases, particularly Evans’ syndrome, this has not been reliably confirmed.2,3 ALPS has been reported among a spectrum of different races. A male predominance has been confirmed by the French ALPS cohort and the National Institutes of Health (NIH) cohort, with the ratio of male to female patients being 2.2 and 1.6 respectively.1,4
About two-thirds of patients with ALPS have an identified genetic defect.5 The germline mutation in the ALPS-FAS gene, also called tumor necrosis factor receptor superfamily member 6, is most commonly implicated. It is inherited in an autosomal dominant manner. Furthermore, about 15% to 20% of patients have somatic mutations in FAS (ALPS-sFAS).6-8 In these patients, somatic mutations are found in alpha/beta double-negative T (DNT) cells and also in other hematopoietic cells.9,10
Leukocyte proliferation and expanded populations result in lymphadenopathy, splenomegaly and, less frequently, hepatomegaly.10 A specific staging system that can be used to predict the prognosis in patients with ALPS has not yet been established. However, prognosis of lymphoproliferation is considered relatively better than autoimmunity, and fewer individuals require long-term treatment with immunomodulating agents such as glucocorticoids, mycophenolate mofetil (MMF), rituximab and high-dose intravenous immune globulin (IVIG).11
The author hereby presents a case of an 11-year-old Asian male, who has been followed up since three years of age for recurrent cytopenias, to raise awareness for early diagnosis and management of ALPS. Written consent was obtained from the patient for the publication of material relating to them in this journal.
An 11-year-old Asian male developed exacerbation of his idiopathic thrombocytopenic purpura, which was diagnosed at age 3 by exclusion. He had presented multiple times to the Georgetown University Hospital with intermittent breakouts of purpuric rash, nosebleeds, and prolonged infections since the time of diagnosis eight years ago. He had no other medical conditions, and his family history was negative for hematological and autoimmune disorders. Vital signs were stable. Successive physical examinations showed mild stable hepatomegaly, and ultrasound examination showed resolving mild splenomegaly. Lymphadenopathy was inappreciable.
Initial workup showed thrombocytopenia and lymphocytopenia. His blood counts over an eight-year follow-up period returned median values of 11.9 g/dL (range: 4.3–14.6 g/dL), 146 × 109/L (range: 2.0–494 × 109/L), and 3.30 × 109/L (range: 0.0005–25.1 × 109/L) for hemoglobin, platelets and absolute neutrophil count, respectively (Table 1). Severity of hemoglobin reached a maximum of grade 3 (<7.0 g/dL) according to Division of Micro-biology and Infectious diseases (DMID) pediatric toxicity tables, and grade 4 for platelets (<25 × 109/L) and ANC (<0.25 × 109/L). A bone marrow analysis returned normal results. The differential diagnosis included common variable immunodeficiency, hyperimmunoglobulin M syndrome, Wiskott-Aldrich syndrome, and hemophagocytic lymphohistiocytosis. Further evaluation showed evidence of positive direct and indirect coombs without hemolysis. Anti-nuclear and antiphospholipid antibody levels were negative, and complement levels were within normal range. Hepatitis B and C serological tests were negative. Antineutrophil antibodies (specific antibodies against the polymorphic human neutrophil antigen) were found to be positive, suggesting an underlying autoimmune neutropenia. Finally, flow cytometry of peripheral blood revealed the presence of double-negative T (DNT) cells (Table 2), solidifying a diagnosis of probable autoimmune lymphoproliferative syndrome (ALPS). Pertinent positive laboratory test results showing the progression of likely diagnoses in this patient can be found in Figure 1.
Table 1.Median, Range and Select Severe Values for Hemoglobin, Platelets and Absolute Neutrophils (ANC) in a Patient with Probable ALPS
| Date [MM.DD.YY] |
Hemoglobin (g/dL) [normal: 13–17] |
Platelets (× 109/L) [normal 150–400] |
Absolute neutrophil count (× 109/L) [normal: 2–8] |
|
|---|---|---|---|---|
| Range | NA | 4.3–14.6 | 2–494 | 0.5–25.1 |
| Median | NA | 11.9 | 146 | 3.3 |
| A | 04.02.08 | 8.7 | 31 | 0.001 |
| B | 06.23.09 | 11.1 | 5 | 0.322 |
| C | 12.03.11 | 11.0 | 4 | 0.609 |
| D | 11.03.12 | 4.3 | 208 | 0.223 |
Lymphocyte Phenotyping Results, Showing Positive Double-Negative T (DNT) Cells in a Patient with Probable ALPS, the Laboratory Marker Unique to the Condition
| Results | Reference range | ||||
|---|---|---|---|---|---|
| % | Abs # (/Ul) | % | Abs # (/Ul) | ||
| Total T cell | CD3 | 72.5 | 2,467 | 58.7–82.6 | 780–2,537 |
| Total B cell | CD20 | 26.4 | 898 | 8.8–27.9 | 118–837 |
| Total NK cell | CD16+ or CD56+/CD3- | 1.3 | 44 | 4.4–28.0 | 70–688 |
| T cell subsets | CD3+/CD4-/CD8- | 10.0 | 340 | 2.4–10.4 | 53–221 |
| DNT/alpha-beta | 3.4 | 116 | 0.4–1.5 | 8–32 | |
| CD8+/CD57+/CD3+ | 333 | 333 | <6.9 | <246 | |
| B cell subsets | CD20/CD5 | 13.3 | 453 | 0.6–10.4 | 16–235 |
| CD20/CD27 | 6.8 | 231 | 1.4–4.9 | 21–111 | |
Pertinent Positive Laboratory Test Results Showing the Progression of Likely Diagnoses in a Patient with Probable ALPS
Relationship of Prednisone Treatment to Hemoglobin, Platelet and Absolute Neutrophil Counts in Patient with Probable ALPS over an Eight-Year Period. A Moving Average Two-period Trend-line has been added (Dotted Line)
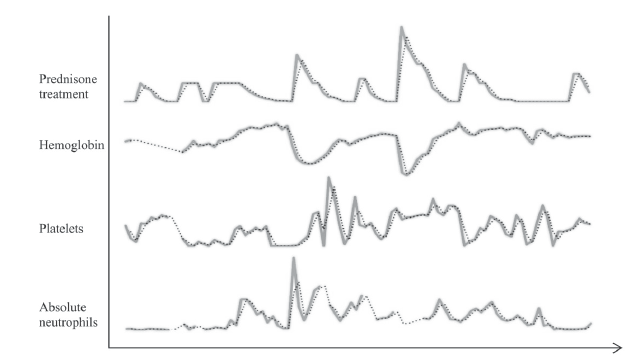
Patient was fully compliant to the treatment. He elicited a favorable response to intravenous immunoglobulin (IVIG) and prednisolone (Figure 2). A trial of rituximab (375 mg/m2), to which he developed fever, chills, and flushing, was not tolerated. Table 1 shows select values at which the patient’s counts were at their lowest; Point D demonstrates severe anemia after he was referred from a nearby emergency center with fever, vomiting, dehydration, and a hemoglobin level of 4.1 g/dL. Upon admission, he was given two units of packed red blood cells, in addition to two doses of IVIG and intravenous methylprednisolone (0.8 mg/kg every 6 hours). His counts began responding after the second transfusion with stabilization and then increase in his hemogloblin to 8.4 g/dL over four days. He was started on 60 mg oral prednisone thrice daily, which was tapered down gradually with continued improvement of symptoms after discharge. As of the writing of this paper, follow-ups have seen the patient’s blood counts and symptoms remain relatively stable with the use of oral prednisone.
The diagnosis of autoimmune lymphoproliferative syndrome (ALPS) is currently based on the presence of two required and six additional criteria.12 Required criteria include the presence of lymphadenopathy and/or splenomegaly, as well as elevated circulating alpha/beta double-negative T (DNT) cells detected using flow cytometric immunophenotyping, considered the hallmark of the disease. These cells are very rare in healthy individuals and other immunological disorders, and typically account for less than 2% of the total lymphocyte count. Other lymphocyte subsets may also be expanded, as well as biomarkers such as vitamin B12, interleukin 10, interleukin 19, and Fas ligand (FAS-L) in serum.13
An abnormal lymphocyte apoptosis assay is no longer considered essential for the diagnosis of ALPS, as patients with both somatic FAS mutations and germline FASL mutations can present with normal apoptosis assays. The routine use of this assay is impractical as it is only offered by very few specialized centers.14,15 A probable ALPS diagnosis can be assumed if both the required criteria and any one of the secondary additional criteria are present.12
Lymphoid precursor proliferation tend to wax and wane for reasons that remain unclear, resulting in various degrees of lymphadenopathy and splenomegaly over the course of the disease.1 Autoimmunity presents in males more often than females, although it may not always be apparent at the time of diagnosis or when lymphoid precursors are in large numbers.
The condition usually involves more than one hematopoeitic cell line, such as combinations of autoimmune hemolytic anemia (AIHA), immune thrombocytopenia (ITP), and less commonly autoimmune neutropenia (AIN). Evans syndrome, the association of autoimmune anemia and immunological thrombocytopenia, can be consistent with ALPS especially with immunophenotyping evidence. In many patients, autoimmunity signifies the transition into ALPS requiring long-term therapy and rendering it a less-favorable prognosis.5
Over the course of the eight-year follow up our patient, he was found to have splenomegaly that fluctuated with time, as well as elevated circulating alpha/beta DNT cells at 3.4% upon lymphocyte phenotyping. The presence of over 3% alpha/beta DNT cells is seldom seen in any conditions other than ALPS.16 Positive direct Coomb’s test, indicating autoimmune hemolysis, and his prior diagnosis with idiopathic thrombocytopenic purpura (ITP) suggested Evans’ syndrome. Furthermore, with his positive antineutrophil antibodies demonstrating autoimmune neutropenia, it was likely that an immunopancytopenia was present.
The aforementioned findings fulfill the revised diagnostic criteria put forward by the First International ALPS workshop in 2009 as a probable diagnosis of ALPS (Table 3). Accordingly, patients with probable ALPS should be treated and monitored in the same way as patients with a definitive diagnosis with immunosuppressive and immunomodulating agents such as glucocorticoids, cyclosporine, rituximab and high-dose intravenous immune globulin (IVIG).10
Table 3.Revised Diagnostic Criteria for ALPS Based on the First International ALPS Workshop of 2009, Showing Criteria Fulfilling a Probable ALPS Diagnosis in this Patient
| Required criteria | Criteria present |
| Chronic (>6 months), nonmalignant, noninfectious lymphadenopathy and/or splenomegaly | Yes |
| Elevated CD3+ TCRαβ+ CD4- CD8- DNT cells (>1.5% of total lymphocytes or >2.5% of CD3+ lymphocytes) in the setting of normal or elevated lymphocyte counts | Yes |
| Additional criteria | Criteria present |
| Primary | |
| Defective lymphocyte apoptosis in 2 separate assays | No |
| Somatic or germline pathogenic mutation in FAS, FASLG, or CASP10 | No |
| Secondary | |
| Elevated plasma sFASL levels, plasma IL-10 levels, serum or plasma vitamin B12 levels or plasma IL-18 levels | No |
| Typical immunohistologic findings as reviewed by a hematopathologist | No |
| Autoimmune cytopenia (hemolytic anemia, thrombocytopenia, or neutropenia) with elevated IgG levels (polyclonal hypergammaglobulinemia) | Yes |
| Family history of a nonmalignant/noninfectious lymphoproliferation with or without autoimmunity | No |
| Definitive diagnosis: Both required criteria plus one primary accessory criterion. | No |
| Probable diagnosis: Both required criteria plus one secondary accessory criterion. | Yes |
Our treatment followed the general recommendations of the American Society of Hematology, which can be broadly applicable to ALPS patients with chronic thrombocytopenia.17 The patient responded greatly to immunosuppression by prednisone therapy. High-dose pulse therapy with intravenous methylprednisolone (≈ 5 mg/kg) followed by low-dose oral prednisone (≈ 1 mg/kg) maintenance was used successfully for the pancytopenias, while tapering over several weeks to avoid the well-documented side effects of corticosteroids. G-CSF therapy (150 micrograms, four times daily) was also shown to be favorable for thrombocytopenia and low ANC. Results with IVIG (1 g/kg) treatment was variable for thrombocytopenia. Rituximab (375 mg/m2) was not tolerated.
Morbidity and mortality in ALPS depend on the severity of the autoimmune disease and development of lymphoma.18 Long-term monitoring and follow-up is necessary to avoid complications associated with the disease and relevant therapy. It is necessary to understand the natural history of this rare disorder to plan for early interventions, such as the use of myocephenolate mofetil (MMF), sirolimus, or stem cell transplantation.19 Finally, the risk-benefit ratio must be assessed before any decision that relates to therapy is commenced.
Key Points:The author would like to thank Amal M. Abu-Ghosh, MD and the kind staff at the Lombardi Comprehensive Cancer Center for their help and guidance during his away rotation at the Medstar Georgetown University Hospital in Washington, DC.
The author has no funding, financial relationships, or conflicts of interest to disclose.
Conceptualization, Data collection, Data analysis and interpretation, Writing, Critical revision of the manuscript, Approval of the final version: YSM.
1. Price S, Shaw PA, Seitz A, Joshi G, Davis J, Niemela JE, et al. Natural history of autoimmune lymphoproliferative syndrome associated with FAS gene mutations. Blood. 2014 Mar 27;123(13):1989–99.
2. Seif AE, Manno CS, Sheen C, Grupp SA, Teachey DT. Identifying autoimmune lymphoproliferative syndrome in children with Evans syndrome: a multi-institutional study. Blood. 2010 Mar 18;115(11):2142–5.
3. Stepensky P, Rensing-Ehl A, Gather R, Revel-Vilk S, Fischer U, Nabhani S, et al. Early-onset Evans syndrome, immunodeficiency, and premature immunosenescence associated with tripeptidyl-peptidase II deficiency. Blood. 2015 Jan 29;125(5):753–61.
4. Neven B, Magerus-Chatinet A, Florkin B, Gobert D, Lambotte O, De Somer L, et al. A survey of 90 patients with autoimmune lymphoproliferative syndrome related to TNFRSF6 mutation. Blood. 2011 Nov 3;118(18):4798–807.
5. Madkaikar M, Mhatre S, Gupta M, Ghosh K. Advances in autoimmune lymphoproliferative syndromes. Eur J Haematol. 2011 Jul;87(1):1–9.
6. Holzelova E, Vonarbourg C, Stolzenberg MC, Arkwright PD, Selz F, Prieur AM, et al. Autoimmune lymphoproliferative syndrome with somatic Fas mutations. N Engl J Med. 2004 Sep 30;351(14):1409–18.
7. Bleesing JJ. Sorting out the causes of ALPS. J Pediatr. 2005 Nov;147(5):571–4.
8. Rossler J, Enders A, Lahr G, Heitger A, Winkler K, Fuchs H, et al. Identical phenotype in patients with somatic and germline CD95 mutations requires a new diagnostic approach to autoimmune lymphoproliferative syndrome. J Pediatr. 2005 Nov;147(5):691–4.
9. Dowdell KC, Niemela JE, Price S, Davis J, Hornung RL, Oliveira JB, et al. Somatic FAS mutations are common in patients with genetically undefined autoimmune lymphoproliferative syndrome. Blood. 2010 Jun 24;115(25):5164–9.
10. Teachey DT, Seif AE, Grupp SA. Advances in the management and understanding of autoimmune lymphoproliferative syndrome (ALPS). Br J Haematol. 2010 Jan;148(2):205–16.
11. Wei A, Cowie T. Rituximab responsive immune thrombocytopenic purpura in an adult with underlying autoimmune lymphoproliferative syndrome due to a splice-site mutation (IVS7+2 T>C) affecting the Fas gene. Eur J Haematol. 2007 Oct;79(4):363–6.
12. Oliveira JB, Bleesing JJ, Dianzani U, Fleisher TA, Jaffe ES, Lenardo MJ, et al. Revised diagnostic criteria and classification for the autoimmune lymphoproliferative syndrome (ALPS): report from the 2009 NIH International Workshop. Blood. 2010 Oct 7;116(14):e35–40.
13. Bleesing JJ. Autoimmune lymphoproliferative syndrome (ALPS). Curr Pharm Des. 2003;9(3):265–78.
14. Siegel RM, Frederiksen JK, Zacharias DA, Chan FK, Johnson M, Lynch D, et al. Fas preassociation required for apoptosis signaling and dominant inhibition by pathogenic mutations. Science. 2000 Jun 30;288(5475):2354–7.
15. Martin DA, Zheng L, Siegel RM, Huang B, Fisher GH, Wang J, et al. Defective CD95/APO-1/Fas signal complex formation in the human autoimmune lymphoproliferative syndrome, type Ia. Proc Natl Acad Sci U S A. 1999 Apr 13;96(8):4552–7.
16. Yeh S, Li Z, Sen HN, Lim WK, Gill F, Perkins K, et al. Scleritis and multiple systemic autoimmune manifestations in chronic natural killer cell lymphocytosis associated with elevated TCRalpha/beta+CD3+CD4-CD8-double-negative T cells. Br J Ophthalmol. 2010 Jun;94(6):748–52.
17. Neunert C, Lim W, Crowther M, Cohen A, Solberg L, Jr., Crowther MA, et al. The American Society of Hematology 2011 evidence-based practice guideline for immune thrombocytopenia. Blood. 2011 Apr 21;117(16):4190–207.
18. Straus SE, Jaffe ES, Puck JM, Dale JK, Elkon KB, Rosen-Wolff A, et al. The development of lymphomas in families with autoimmune lymphoproliferative syndrome with germline Fas mutations and defective lymphocyte apoptosis. Blood. 2001 Jul 1;98(1):194–200.
19. Rao VK, Dugan F, Dale JK, Davis J, Tretler J, Hurley JK, et al. Use of mycophenolate mofetil for chronic, refractory immune cytopenias in children with autoimmune lymphoproliferative syndrome. Br J Haematol. 2005 May;129(4):534–8.
Yazan S. Mousa, 1 Faculty of Medicine, Jordan University of Science and Technology, Irbid, Jordan.
Correspondence Yazan S. Mousa. Address: Faculty of Medicine, Jordan University of Science and Technology, Ar Ramtha, Irbid, Jordan. Email: ysmousa09@med.just.edu.jo
Cite as: Mousa YS. Autoimmune lymphoproliferative syndrome (ALPS): a case report. Int J Med Students. 2016 Sep-Dec;4(3):123-6.
Copyright © 2016 Yazan S. Mousa
International Journal of Medical Students, VOLUME 4, NUMBER 3, December 2016