Reviews
Vascular Anomalies Review of the Head and Neck for Physicians in Training
Caleb M. Allred1, Kaitlyn B. Zenner2, Juliana Bonilla-Velez3
doi: http://dx.doi.org/ijms.2024.2665
Volume 12, Number 3: 284-293
Received 22 04 2024;
Rev-request 08 05 2024;
Rev-recd 08 06 2024;
Accepted 08 08 2024
ABSTRACT
A basic understanding of vascular anomalies will aid physicians-in-training as they
seek to properly diagnose and determine interventions for these patient presentations.
The aim of this review is to create a resource for physicians in training that encompasses
the most important clinical aspects of vascular anomalies. Vascular anomalies of the
head and neck are divided into two categories: vascular tumors and vascular malformations.
This review will first describe vascular tumors followed by vascular malformations
and discuss major pathology found in both categories of vascular anomaly. The MEDLINE/PubMed
database was searched for primary research and reviews discussing various vascular
anomalies which include infantile hemangioma, congenital hemangioma, pyogenic granuloma,
tufted angioma, kaposiform hemangioendothelioma, capillary malformations, lymphatic
malformations, venous malformations, and arteriovenous malformations. We conducted
the search from July 5, 2023, to March 21, 2024. Vascular anomalies are frequently
found in pediatric populations and can persist into adulthood, making it important
for trainees to identify them on physical exam. This developing field seeks to improve
form, function, and quality of life for patients with vascular anomalies and often
requires a multidisciplinary approach (i.e., otolaryngology, dermatology, genetics,
plastic surgery, interventional radiology). Various medical and surgical treatment
options are available. A basic knowledge of these anomalies will allow for accurate,
early diagnosis and appropriate intervention which can ultimately improve patient
outcomes.
Introduction
Vascular anomalies are diseases that involve abnormal development of blood vessels.
Further understanding of their development and behavior have helped us differentiate
these diagnoses into vascular tumors and vascular malformations.3 Proper categorization of these diagnoses informs treatment, helps to identify potential
complications, and aids patients' families to understand the natural course of these
conditions. One of the most common vascular anomalies, infantile hemangioma, has been
found to have an incidence as high as 4.5%,1 making it crucial for physicians-in-training to recognize and understand the management
of its pathology. While various articles and textbooks2 intended for physicians are available to study vascular anomalies, a review of the
common pathologies, their diagnosis and treatment targeted toward trainees does not
yet exist. The purpose of this review is to provide a resource for physicians-in-training
seeking to aid their patients in diagnosis and treatment decision-making. To achieve
an appropriate diagnosis, trainees should recognize various pitfalls that may present.
For example, vascular tumors can be mistaken for other tumors or infections. Additionally,
different imaging modalities may show varying degrees of malformation that can be
misinterpreted by radiologists. Therefore, an adequate history and physical exam should
be performed to ensure a comprehensive understanding of the malformation.
Vascular Tumors
Vascular tumors are neoplasms originating from vessels that impact vessel organization
and development. They are classified based on their malignant or local destruction
potentials.Click or tap here to enter text.4 The following is a description of the epidemiology, pathophysiology, diagnosis, and
management of some of the common vascular tumors.
Infantile Hemangioma
Epidemiology
Infantile hemangioma (IH) is the most commonly diagnosed soft tissue tumor and is
present in 4–10% of children.5 Though not typically present at birth, they generally become clinically apparent
within the first month of life and continue to proliferate until 3–4 months of age.5 Growth of IH ultimately plateaus, followed by an involution phase. Generally, 50%
involution occurs by age 5 and 70% by age 7. Involution is often incomplete and can
cause permanent skin changes or disfigurement.Click or tap here to enter text.5–7 The most common risk factors for IH include preterm birth, placental abnormalities,
female gender, low birth weight, being a product of multiple gestations, and family
history.8
Pathophysiology
Although the pathophysiology of IH is unclear, growing evidence suggests a placental
origin due to multiple similarities including growth pattern (9 months of rapid growth)
and presence of similar molecular markers (GLUT1, transcriptome, microRNA profiles).2,9–11
Diagnosis
The most common location for IH to present is the head and neck, potentially impacting
aesthetics, and functionality.12 Clinical appearance varies broadly depending on location, depth, extent, and stage
of evolution.5 IH are classified by depth: superficial, mixed, or deep. Superficial IH present as
bright red plaques, nodules, or masses. Deep IH appear as skin-colored masses with
a bluish tint (Figure 1, A-C). Mixed IH are a combination of bright plaques and skin-colored masses.Click or tap
here to enter text.13 An alternative classification is employed based on extent: IH arising from a single
growth focus are categorized as localized/focal, while segmental IH presents in a
linear or geographic cutaneous area.13,14 IH is primarily diagnosed via history and physical exam. Imaging is used when uncertainties
arise (i.e., distinguishing among vascular anomalies from more aggressive neoplasia,
to delineate the extent of the tumor).115 Generally, ultrasonography with doppler is the first modality of choice. An IH in
the proliferative phase appears as a well-defined mass with non-homogeneous echostructure
that demonstrates high-flow uniform vessel distribution.16 While the mass is involuting, it will appear more hyperechoic due to the increased
fat deposition and present with less vascular density.16 MRI is helpful when determining depth of involvement or in cases of visceral involvement.
IH are well-circumscribed masses that are isointense on T1-weighted sequences, hyperintense
on T2-weighted sequences, and show avid post-contrast enhancement without arteriovenous
shunting (as seen in arteriovenous malformations).17
Figure 1.
Infantile Hemangiomas
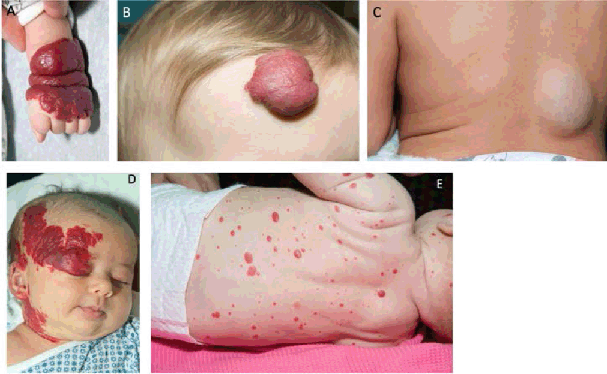
Legend: A. Segmental upper extremity IH. B. Superficial hemangioma of the forehead. C. Deep
hemangioma of the back. D. Segmental facial hemangioma, concerning for PHACES.2 E. Hemangiomatosis. Multiple infantile hemangiomas (≥5) place patients at increased
risk of hepatic hemangioma.
Management and Prevention
It is important to keep in mind that the most common treatment for hemangiomas is
observation, as they commonly undergo involution. Treatment is reserved for complicated
and high risk IH cases. Complications are typically seen during the proliferative
phase for large IH located on the face including bleeding, airway compromise, visual
compromise, ulceration, pain, difficulty feeding or risk permanent disfigurement.18 See Table 1 for special clinical considerations.
Table 1.
Special Considerations for Infantile Hemangiomas.
| Clinical Presentation |
Concern |
Workup |
| Hemangiomatosis (>5) |
Screen for intrahepatic/visceral hemangiomas |
Abdominal ultrasound.82
Consider thyroid function tests in multifocal and diffuse hepatic hemangiomas.7 |
| Segmental facial hemangioma |
Rule out PHACES syndrome [posterior fossa brain malformations, segmental cervicofacial
hemangioma, arterial abnormalities (vessel abnormalities in the head or neck), cardiac
abnormalities or aortic coarctation, eye abnormalities, sternal clefting]83 |
Ophthalmology referral.
Echocardiogram, cardiology referral.
MRI/MRA of head, neck, and arch.84 |
| Midline lumbosacral hemangiomas |
Rule out SACRAL syndrome (spinal dysraphism, anogenital anomalies, urogenital anomalies). |
Referral to dermatology urology, nephrology, neurology, and/or neurosurgery based
on clinical needs.
|
| Large hemangiomas, particularly in the liver |
Risk of high-output cardiac failure and hypothyroidism. |
Cardiology evaluation and TFT if concern. |
| Beard distribution segmental hemangioma (facial lower third and neck) or central
neck
|
Rule out airway hemangiomas.85 |
Otolaryngology evaluation to consider airway evaluation. |
| Ulceration and bleeding |
|
|
| Perineal, axilla, neck |
Risk of ulceration related to friction. |
Monitor clinically and treat as needed. Treatment can include topical antibiotic ointment,
wound care, and on occasions culture and oral antibiotics.
|
| Compromise vital functions |
|
|
| Periorbital |
Can cause astigmatism, strabismus, or amblyopia. |
Ophthalmology referral. |
| Perioral, lip |
Feeding difficulties. |
Otolaryngology evaluation and feeding therapy consultation if appropriate. |
| Airway |
Becoming symptomatic (stridor, feeding difficulties, etc.) and can become life-threatening
if airway obstruction progresses.
|
Otolaryngology evaluation for airway assessment. |
| Nasal |
Nasal obstruction. |
Otolaryngology evaluation for airway assessment. |
| Slow Involution or Deformity |
|
|
| Parotid |
Can be deep and without cutaneous manifestations. |
Ultrasound can aid in diagnosis.86
Risk to the facial nerve during surgery. Recommend facial nerve monitoring ±mapping.87 |
| Cosmetic Concern |
|
|
| Nasal tip, ear, large facial |
Social impact from facial disfigurement. |
Lower threshold to treat. |
First line therapy for IH is oral propranolol, a nonselective beta-adrenergic blocker.19 Contraindications to propranolol treatment should be considered including cardiogenic
shock, sinus bradycardia, heart failure, bronchial asthma, allergy, and history of
hypoglycemia. However, these conditions are all uncommon in infants. Dosing of the
propranolol for IH is between 1 and 3 mg/kg/day separated into 2–3 doses with 6 hours
between doses. Cardiac consult should be obtained before therapy initiation if there
is concern for cardiac disease. Patients should be monitored for side effects including
bradycardia, hypoglycemia, hypotension, and bronchospasm. These are not common and
typically not life threatening.19,20 Historically, corticosteroids were the first-line medical therapy for IH. Intralesional
corticosteroids can still be used to decrease the size of small IH, but propranolol
is preferred.21 Another treatment option is pulsed dye laser (PDL). This technique can be used for
ulcerated IH or residual visible vascularity after IH involution. Finally, surgical
excision may be performed for patients who cannot undergo systemic therapy. Additionally,
surgery may be performed if propranolol is ineffective, for cosmetic deformity, or
caregiver preference.2 See Table 2 for a summary of indications for treatment of IH.
Table 2.
Indications for Treatment of Infantile Hemangiomas.
| Indication |
Locations to consider |
Special considerations |
| Compromise vital functions |
Periorbital |
Can cause astigmatism, strabismus, or deprivation amblyopia.
Consider ophthalmology referral. |
|
Perioral, lip |
Can result in feeding difficulties.
Consider feeding therapy and/or otolaryngology evaluation. |
|
Airway |
Can present with stridor or feeding difficulties and may become life threatening.
Recommend otolaryngology evaluation.
|
|
Nasal |
Can disrupt nasal airflow in obligate nasal breathers. Consider otolaryngology evaluation. |
| Slow Involution or Deformity |
Parotid |
Can be deep and without cutaneous manifestations so ultrasound aids in diagnosis.80
Risk to the facial nerve during surgery. Recommend facial nerve monitoring +/− mapping.
81 |
| Cosmetic Concern |
Nasal tip, ear, large facial |
Can result in permanent disfigurement. |
Segmental IH and Syndromes
PHACES Syndrome
PHACES syndrome stands for posterior fossa intracranial abnormalities, hemangiomas, arterial abnormalities, cardiac defects and coarctation of the aorta, eye abnormalities, and sternal clefting.Click or tap here to enter text.22 This syndrome is associated with cervicofacial IH (Figure 1C). Patients with PHACES are more likely to have an airway IH and abnormal cerebral
vasculature, a risk factor for stroke.23
SACRAL and PELVIS Syndromes
PELVIS is another syndrome associated with hemangiomas that stands for perineal hemangiomas, external genital malformations, lipomyelomeningocele, vesicorenal abnormalities, imperforate anus, or skin tags.24 Furthermore, SACRAL syndrome is associated with angiomas in the lumbosacral region
and stands for spinal dysraphism with anogenital, cutaneous, renal, and urologic anomalies, associated with angiomas in the lumbosacral region.25
Hemangiomatosis
Infants with five or more cutaneous IH (Figure 1E) have an increased risk of hepatic IH which prompts screening imaging via abdominal
ultrasound.26 Though often clinically benign, hepatic IH can be complicated by bleeding, congestive
cardiac failure, hypothyroidism, and abdominal compartment syndrome.Click or tap here
to enter text.27 Hepatic IH generally undergo involution similar to cutaneous IH.27
Congenital Hemangioma
In contrast to IH, congenital hemangiomas are present at birth (Figure 2A) and are GLUT-1 negative despite having similar histology.28 Congenital hemangiomas are divided into two subtypes: rapidly involuting congenital
hemangioma (RICH) and non-involuting congenital hemangioma (NICH). RICH will resolve
over the first year of life while NICH do not generally resolve spontaneously.Click
or tap here to enter text.29 Thus, NICH can treated with surgery or laser.
Figure 2.
Vascular Tumors.
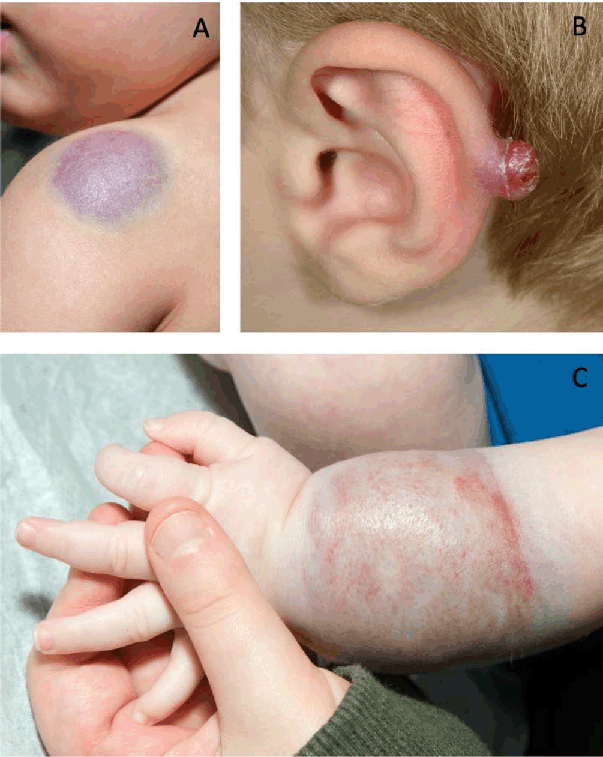
Legend: A. Congenital hemangioma. Blue-purple deep mass with surrounding pale halo. B. Pyogenic
granuloma.2 Pedunculated, erythematous growth that commonly ulcerates and presents with bleeding.
C. Kaposiform hemangioendothelioma. Violaceous skin discoloration with palpable mass
underlying.
Pyogenic Granuloma
This benign vascular tumor, also known as lobular capillary hemangioma, can arise
in both children and adults (Figure 2B). Children will generally develop pyogenic granuloma in the head and neck region
while adults often develop lesions on the trunk.Click or tap here to enter text.30,31 Mucosal pyogenic granulomas also occur in about 2% of pregnancies between the second
and fifth months.Click or tap here to enter text.32 These tumors are rapidly growing, exophytic, red-colored papules that bleed commonly.
Similar to congenital hemangioma, they are GLUT-1 negative on histopathology. The
treatment is surgical, either by complete excision or punch excision with curettage
or electrodesiccation of the feeding vascular stalk.33
Kaposiform Hemangioendothelioma And Tufted Angioma
Both tufted angioma (TA) and kaposiform hemangioendothelioma (KHE) are vascular tumors
with a lymphatic component. TA is localized and noninvasive, however KHE invades nearby
tissue.Click or tap here to enter text.34 KHE presents clinically as violaceous nodules and demonstrates tissue invasion on
imaging (Figure 2C). TA present similarly, may or may not involve skin, and do not invade local tissue.
Both of these conditions are associated with the Kasabach-Meritt phenomenon (KMP),
a consumptive coagulopathy characterized by thrombocytopenia, hypofibrinogenemia,
and anemia.Click or tap here to enter text.35 Surgery and medical therapy (e.g., sirolimus, vincristine) can be employed for treatment
of both conditions.36
Vascular Malformations
Vascular malformations occur due to errors in the initial development of blood and
lymph vessels and tend to be present at birth and grow with the child.37 Vascular malformations can present with complex clinical presentations and management.37 See Table 3 for a summary of vascular malformations by common complications and treatment.
Table 3.
Indications for Treatment of Infantile Hemangiomas.
| Vascular malformation |
Complications |
Treatment |
| Capillary malformations |
Generally cosmetic (see Figure 3A) can topical sirolimus. be associated with Beckwith-Wiedemann, Nova, macrocephaly-capillary
malformation, and Sturge Weber syndromes. 9, 36 |
Pulsed dye laser and topical sirolimus.9 |
| Lymphatic malformations |
Impaired swallowing function, airway compromise, cosmetic.42, 43 Can be associated with CLOVES and KTS syndromes.48 |
Many resolve 45 spontaneously. Sclerotherapy, surgical excision, medications (sirolimus; aspirin
and PI3K inhibitors under investigation). 51 |
| Venous malformations |
Pain and localized intravascular coagulopathy which may predispose to disseminated
intravascular coagulopathy. Associated
with Klippel-Trenaunay syndrome.54 |
Perioperative treatment with low molecular weight heparin (LMWH) and surgery. Medical
treatments include aspirin and sirolimus. Compression may be helpful with extremity
presentations.62 |
| Arteriovenous malformations |
Deformity, bleeding, and high output cardiac failure. Other complications include
infiltration of surrounding tissues and increased size during pregnancy, puberty,
and trauma.9,65 |
Medical management with doxycycline, sirolimus, and trametinib for severe cases. Intralesional
bleomycin has shown variable results. Other options include ablation of draining veins
with flashlamp-pumped pulsed dye laser (FPDL) and Nd:YAG laser. Surgical resection
for focal AVM. 9, 72, 73 |
Capillary Malformations
The most common vascular malformations involve capillaries. Capillary malformations
(CM) (Figure 3A) encompass a wide range of cutaneous malformations linked by their abnormal capillary
morphology on histology.
Figure 3.
Vascular Malformations.
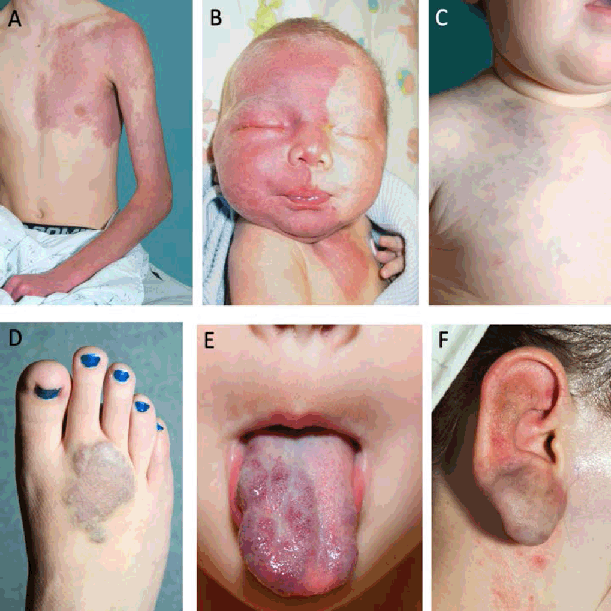
Legend: A. Capillary malformation of the left upper extremity and chest. B. Segmental facial
port wine stain (PWS) associated with Sturge Weber syndrome (SWS).2 Venous malformations. C-E: cutaneous presentation of venous malformations of the
chest (C) and foot (D) seen as violaceous discoloration of the skin. E: mucosal venous
malformation of the tongue. F. Arteriovenous malformations of the ear.
Nevus simplex
This CM, commonly found in Caucasian infants, Click or tap here to enter text.38 has a wide range of names including salmon patch, angel's kiss (when located on the
glabella), and stork bite (when located on the nape of the neck). These blanching
erythematous macules present with indistinct borders and typically resolve over months
to years. They are occasionally linked with Beckwith-Wiedmann syndrome, Nova syndrome,
and macrocephaly-capillary malformation syndrome (MCAP).2
Port Wine Stain and Sturge Weber Syndrome
One CM that does not resolve is the port wine stain (PWS), also known as nevus flammeus.
PWS will grow in conjunction with the child and present as pink to dark red patches
with well-demarcated borders (Figure 3B). Also, it may thicken or become darker over time and is associated with soft tissue
and bone hypertrophy, gingival hyperplasia, and dental anomalies.39 Around 10% of patients with facial PWS have Sturge-Weber syndrome (SWS).Click or
tap here to enter text.40 SWS is the association of facial CM with leptomeningeal angiomatosis and glaucoma.
Some patients with SWS may develop epilepsy. MRI with contrast is the gold standard
for diagnosis.41
Telangiectasias and hereditary hemorrhagic telangiectasia
Telangiectasias are small, dilated capillary vessels that present as red macules with
stellate shape and a pale halo. Isolated, they are nonsyndromic; however, multiple
telangiectasias together may indicate hereditary hemorrhagic telangiectasia (HHT).
This autosomal dominant disorder presents with telangiectasias affecting multiple
systems, arteriovenous malformations (AVM), and vascular dysplasia. This disorder
most commonly presents at age 12 and its severity increases with age. HHT can lead
to life-threatening bleeding.42
Treatment
CM treatment is tailored to each individual presentation and the needs of the patient.
Generally, a balance is sought between intervening before the lesion thickens and
becomes nodular, while minimizing risks of anesthetics (generally after 6 months of
age).2 First-line treatment for CM is pulsed dye laser with or without topical sirolimus.43,44
Lymphatic Malformations
Lymphatic malformations (LM) are a subset of vascular malformations that present with
abnormal lymphatic vessel morphology. LM occur in approximately 1 in 4000 live births.
Prior nomenclature used to describe LM included cystic hygroma and lymphangioma, however
these terms imply a watery tumor rather than a collection of ill-formed lymphatic
vessels and are no longer in use. Some LM such as posterior cervical LM or extensive
cervicofacial LM can be diagnosed prenatally. Posterior cervical LM are sometimes
associated with syndromic conditions and regress by birth.45 Ventral, isolated LM can also be diagnosed prenatally or shortly after birth and
commonly present in the neck and face. In utero, LM may be associated with polyhydramnios
if swallowing function is impaired and/or may have airway compromise at birth. A LM
with airway obstruction requires delivery planning and a possible EXIT (ex-utero intrapartum
treatment) procedure.46,47
Diagnosis and classification
LM are diagnosed via a combination of history, physical exam, and imaging. Depending
on the extent of the malformation, MRI and CT are commonly used modalities that can
aid in the diagnosis of LM and selection is based on preference. The authors prefer
MRI given the better soft tissue delineation and avoidance of radiation in children.
If radiographic cystic spaces in the LM are >2cm in diameter they are classified as
macrocystic. Those less than 2cm in diameter are classified as microcystic. LM that
presents in the head and neck are classified using de Serres stages, which is based
on laterality and location relative to the hyoid.48 More than 80% of LM are stage I-III with unilateral involvement.
Generally, unilateral LM do not cause functional compromise and can resolve spontaneously.49 Large, bilateral LM typically cause functional compromise and do not resolve spontaneously.49 LM are not hereditary, rather, they are associated with somatic mutations in PIK3CA, an oncogene associated with cancer and other overgrowth disorders.50,51
Associated syndromes: CLOVES and KTS
LM are not generally associated with a syndrome and present in isolation but may be
associated with other PIK3CA-related overgrowth syndromes (PROS). PROS associated with LM include CLOVES syndrome
(congenital lipomatosis, overgrowth, vascular malformations, epidermal nevi, and skeletal
anomalies) and Klippel-Trenaunay syndrome (KTS) (port wine stains, venous malformations,
LM, and overgrowth).52 Overgrowth in CLOVES syndrome can be very severe and generally involves multiple
limbs and possibly trunk. Klippel-Trenaunay syndrome generally only has overgrowth
of a single extremity.53
Management and Prevention
Treatment of LM is typically pursued in response to functional or aesthetic compromise.
Many resolve spontaneously, especially unilateral macrocystic LM.49 If infected, it typically responds well to systemic antibiotics and corticosteroids.
Another treatment that may be pursued for macrocystic LM is sclerotherapy, a procedure
in which interventional radiology drains the associated fluid and injects a sclerosing
agent.54 This procedure is often required multiple times to achieve a sustained result. Surgical
excision is also an option. The most commonly used medication for LM is sirolimus,
an MTOR inhibitor. Aspirin and targeted PI3K inhibitors (alpelisib) are undergoing
investigation as potential treatments for LM.55
Venous Malformations
Epidemiology
Around 1–4% of individuals have a venous malformation (VeM). VeM commonly present
in the head and neck, generally on mucosal surfaces or within muscles.Click or tap
here to enter text.56 The next most common area for a VeM to arise is the upper and lower extremities.
VeM often clinically present at puberty, but most are present even at birth and grow
proportionately with the child.Click or tap here to enter text.57 VeM are visualized as a mass with a bluish hue overlying the skin but may also present
without the blue hue (Figure 3C-E). Mucosal VeM typically presents with blue-purple discoloration.
Diagnosis
A combination of clinical history, physical exam, and imaging is used for diagnosis.
Choosing the imaging modality depends on the location, patient age, and ability to
receive contrast. Ultrasound, CT, and MRI may all provide valuable information. Many
VeM present with pain and there may also be localized intravascular coagulopathy (LIC)
which can occasionally predispose to disseminated intravascular coagulopathy (DIC).
LIC is most commonly seen in large lesions (>10 mL), when phleboliths are present,
in multifocal disease, and when associated with Klippel-Trenaunay syndrome.58 Elevated D-dimer levels in a patient with VeM has a high specificity (>97%) for LIC.59
Etiology and associated syndromes
VeM can arise sporadically or hereditarily. Various mutations are linked with VeM:
sporadic venous malformations are associated with somatic mutations in TIE2/TEK and PIK3CA,60,61 some familial VeM are associated with mutations in RASA1,62 and hereditary venous malformations, also known as familial venous malformation cutaneo-mucosal
(VMCM), are associated with inherited mutations in TIE2/TEK.63 Glomuvenous malformation is a hereditary disease associated with inherited mutations
in GLMN (glomulin) with a distinct pathologic phenotype that involves glomus cells, a type
of immature vascular smooth muscle cell.64 Blue rubber bleb nevus syndrome is a form of VeM that presents with compressible
mucocutaneous VeM and involves the visceral organs, most commonly in the gastrointestinal
tract.65
Management and Prevention
Patients with bothersome VeM or who present with LIC are candidates for treatment.
A patient with VeM and elevated D dimer should be treated perioperatively with LMWH
for any sedated procedure as well as 14 days pre and post procedure.66 Medical therapies include aspirin and sirolimus. For patients with VeM on the extremity,
compression can help to relieve symptoms though it is not known whether it helps to
prevent LIC.
VeM often requires invasive therapy such as laser therapy, sclerotherapy, and surgery.
Laser therapy can include PDL or Nd:YAG for superficial lesions. Sclerotherapy is
also available but often requires multiple procedures to achieve an effect.67 Surgical excision can be performed, often with glue embolization in which interventional
radiology injects the VeM with n-BCA glue immediately prior to resection to decrease
the risk of bleeding and allow for more thorough resection.68
Arteriovenous Malformations
Epidemiology
Arteriovenous malformations (AVM) involve atypical connections between arteries and
veins (Figure 3F). AVM have a central nidus, one or more arteries that feed the nidus, and one or
more veins that drain it. AVM can be intracranial or extracranial. When extracranial
they are most likely to occur in the head and neck. AVM are present at birth but commonly
increase in size during pregnancy, puberty, or trauma.Click or tap here to enter text.2,69 Contrary to other vascular malformations, AVM commonly infiltrate adjacent tissue.
Common complications include deformity, bleeding, and high output cardiac failure
if large in size. If extracranial, the Schobinger classification can categorize AVM
based on presentation and exam. Intracranial AVM are classified based on size and
morphology with the Spetzler-Martin scale.Click or tap here to enter text.70 Imaging is helpful for diagnosis and requires arteriography or MR-angiogram. Smaller
AVM with a single feeding artery may be treatable, but effective therapeutic treatments
for diffuse AVM are more challenging.
Etiology and associated syndromes
AVM are generally sporadic and can be associated with somatic mutations in KRAS, BRAF and MAP2K1.Click or tap here to enter text.71–73 Some forms of AVM can be inherited. For example, CM-AVM is an autosomal dominant
disorder, commonly associated with mutations in RASA1, that presents with multiple
round to oval vascular stains with a vasoconstrictive halo.62 Around one third of individuals with a CM-AVM also have an AVM in the brain, spine,
bone, skin, or soft tissue.74 One subtype of CM-AVM is Parkes-Weber syndrome which can present with arteriovenous
fistula (AVF) and lower extremity limb overgrowth. Hereditary hemorrhagic telangiectasia
(HHT), a syndrome associated with mutations in the TGF-beta pathway, is most commonly
associated with pulmonary AVM, but may also present with gastrointestinal tract, liver,
spine, or brain AVM.75
Treatment
AVM differ from VeM and LM in that they should not be observed. They warrant aggressive
therapy given their propensity to infiltrate surrounding tissue. However, functional
and aesthetic outcomes associated with treatment must be taken into consideration.
Medical management has seen some response with doxycycline (matrix metalloproteinase
inhibitor), sirolimus (MTOR inhibitor), and trametinib (MAP2K1 inhibitor) for severe
AVM.76,77 Intralesional bleomycin has also been trialed with variable results. Other options
include flashlamp-pumped pulsed dye laser (FPDL) and Nd:YAG laser to ablate superficial
draining veins.2 The primary treatment of AVM is embolization and surgical resection. Embolization
can be done with various agents including onyx, ethanol, and n-BCA (glue). Onyx and
n-BCA embolization are generally used as an adjunct to surgery in both intracranial
and extracranial AVM.78 Surgical resection is most successful in focal AVM and is generally performed after
or in conjunction with embolization.79
Discussion
This review of vascular anomalies has discussed the pathogenesis, diagnosis, and management
of the two main categories: vascular tumors and vascular malformations. Vascular anomalies
are prevelant5 and recognizing them clinically and understanding management will aid trainees in
their clinical efforts. Various reviews of vascular anomalies have previously been
published. For example, a brief review by Cox et. alClick or tap here to enter text.80 organized vascular anomalies by subtype to help clinicians develop a clear understanding
of the clinical aspects, diagnostic tools, imaging modalities, and options for interventions
available. More extensive reviews, such as Perkins and Balakrishnan,2 have been developed to provide a comprehensive resource of current evidence-based
management of head and neck vascular anomalies for providers. The purpose of this
review was to provide an educational tool for physicians-in-training seeking to gain
an understanding and assist in the recognition of these pathologies, as well as to
offer an insight into the various treatment methods.
This review has several strengths, including a concise delineation of common vascular
anomalyies, visual images to support recognition, and its originality as the first
vascular anomalies review with physicians-in-training as the target audience. Limitations,
however, do exist. Our understanding of the epidemiology of vascular anomalies continues
to develop and studies evaluating the effectiveness of treatment options continue
to be published. Thus, our understanding and explanations of certain vascular anomalies
is extensive, while less is known about more recently identified pathologies. This
understanding will continue to develop as future research will likely focus on the
refinement of genetic and molecular therapies, improved diagnostic techniques, and
the development of personalized treatment protocols.3,19,81 Future reviews of vascular anomalies should continue to be published in an effort
to provide up-to-date resources for this developing field.
Conclusion
This review seeks to describe the epidemiology, diagnosis, and treatment of the most
common vascular anomalies. It is intended for physicians-in-training seeking an up-to-date
resource for understanding and managing vascular anomalies that is appropriate to
their training level. As medical school curricula vary in the depth of coverage of
vascular anomalies, this review will allow students to have a comprehensive, trainee-level
guide. As studies continue to further our knowledge of these pathologies, updated
reviews should continue to be published to provide up-to-date resources for trainees
and clinicians.
Summary – Accelerating Translation
Title: Vascular Anomalies Review of the Head and Neck for Physicians in Training
The main problem to solve: Limited training on vascular anomalies of the head and
neck is given to medical students. However, a basic understanding of vascular anomalies
can aid physicians-in-training as they seek to properly diagnose and determine interventions
for these patient presentations.
Aim of this review: To create a resource for physicians in training that encompasses
the most important clinical aspects of vascular anomalies.
Methodology: Current evaluation, diagnosis, and treatment for vascular tumors and
vascular malformations were compiled into a trainee-level resource through literature
review.
Results: Current evaluation, diagnosis, and treatment guidelines were described for
infantile hemangioma, segmental IH and syndromes, pyogenic granuloma, kaposiform hemangioendothelioma
and tufted angioma, capillary malformations, lymphatic malformations, venous malformations,
and arteriovenous malformations.
Conclusion: A basic knowledge of these anomalies will allow students to assist in
accurate, early diagnosis and appropriate intervention of vascular anomalies. This
can ultimately improve patient outcomes.
Acknowledgments
All clinical photos were published courtesy of the Seattle Children's Vascular Anomalies
Program, Jonathan A. Perkins, and Eden Palmer.
Conflict of Interest Statement & Funding
The National Institute on Deafness and Other Communication Disorders supports Kaitlyn
Zenner via the University of Washington Otolaryngology Research Training Program (PI:
Dr. Jennifer Stone, PhD; Grant Number: 2T32000018).
Author Contributions
Conceptualization: CMA, KBZ, JBV. Writing – Original Draft: CMA, KBZ, JBV. Writing
– Review Editing: CMA, KBZ, JBV.
References
1. Munden A, Butschek R, Tom WL, Marshal JS, Poeltler DM, Krohne SE, et al Prospective study of infantile haemangiomas: incidence, clinical characteristics and
association with placental anomalies. Br J Dermatol. 2014;170(4):907–913.
2. Perkins JA, Balakrishnan K. Evidence-Based Management of Head and Neck Vascular Anomalies. (Springer International Publishing, ed.).; 2018.
3. Wassef M, Blei F, Adams D, Alomari A, Baselga E, Berenstein A, et al Vascular Anomalies Classification: Recommendations From the International Society
for the Study of Vascular Anomalies. Pediatrics. 2015;136(1):e203–14.
4. Mansfield SA, Williams RF, Iacobas I. Vascular tumors. Semin Pediatr Surg. 2020;29(5):150975.
5. Drolet BA, Esterly NB, Frieden IJ. Hemangiomas in children. N Engl J Med. 1999;341(3):173–181. 10.1056/NEJM199907153410307
6. Leung AKC, Lam JM, Leong KF, Hon KL. Infantile Hemangioma: An Updated Review. Curr Pediatr Rev. 2021;17(1):55–69.
7. DeHart A, Richter G. Hemangioma: Recent Advances. F1000Res. 2019; 8.
8. Smith CJF, Friedlander SF, Guma M, Kavanaugh A, Chambers CD. Infantile Hemangiomas: An Updated Review on Risk Factors, Pathogenesis, and Treatment. Birth Defects Res. 2017;109(11):809–815.
9. North PE, Waner M, Mizeracki A, Mihm MC. GLUT1: a newly discovered immunohistochemical marker for juvenile hemangiomas. Hum Pathol. 2000;31(1):11–22.
10. Barnés CM, Huang S, Kaipainen A, Sanoudou D, Chen E, Eichler, G, et al Evidence by molecular profiling for a placental origin of infantile hemangioma. Proc Natl Acad Sci U S A. 2005;102(52):19097–19102.
11. Strub GM, Kirsh AL, Whipple ME, Kuo W, Keller R, Kapur R, et al Endothelial and circulating C19MC microRNAs are biomarkers of infantile hemangioma. JCI Insight. 2016;1(14):e88856.
12. Huoh KC, Rosbe KW. Infantile hemangiomas of the head and neck. Pediatr Clin North Am. 2013;60(4):937–949.
13. Chiller KG, Passaro D, Frieden IJ. Hemangiomas of infancy: clinical characteristics, morphologic subtypes, and their
relationship to race, ethnicity, and sex. Arch Dermatol. 2002;138(12):1567–1576.
14. Haggstrom AN, Lammer EJ, Schneider RA, Marcucio R, Frieden IJ. Patterns of infantile hemangiomas: new clues to hemangioma pathogenesis and embryonic
facial development. Pediatrics. 2006;117(3):698–703.
15. Darrow DH, Greene AK, Mancini AJ, Nopper AJ, SECTION ON DERMATOLOGY SOOANS and SOPS. Diagnosis and Management of Infantile Hemangioma. Pediatrics. 2015;136(4):e1060–104.
16. Esposito F, Ferrara D, Di Serafino M, Severino M, Nosari R, Galdieri M, et al Classification and ultrasound findings of vascular anomalies in pediatric age: the
essential. J Ultrasound. 2019;22(1):13–25.
17. Moukaddam H, Pollak J, Haims AH. MRI characteristics and classification of peripheral vascular malformations and tumors. Skeletal Radiol. 2009;38(6):535–547.
18. Haggstrom AN, Drolet BA, Baselga E, Chamlin S, Garzon M, Horii K, et al Prospective study of infantile hemangiomas: clinical characteristics predicting complications
and treatment. Pediatrics. 2006;118(3):882–887.
19. Drolet BA, Frommelt PC, Chamlin SL, Haggstrom A, Bauman N, Chiu R, et al Initiation and use of propranolol for infantile hemangioma: report of a consensus
conference. Pediatrics. 2013;131(1):128–140.
20. Hoeger PH, Harper JI, Baselga E, Bonnet D, Boon LM, Atti MCD, et al Treatment of infantile haemangiomas: recommendations of a European expert group. Eur J Pediatr. 2015;174(7):855–865.
21. Barrio VR, Drolet BA. Treatment of hemangiomas of infancy. Dermatol Ther. 2005;18(2):151–159.
22. Frieden IJ, Reese V, Cohen D. PHACE syndrome. The association of posterior fossa brain malformations, hemangiomas, arterial anomalies,
coarctation of the aorta and cardiac defects, and eye abnormalities. Arch Dermatol. 1996;132(3):307–311.
23. Hess CP, Fullerton HJ, Metry DW, Drolet B, Siegel D, Auguste KI, et al Cervical and intracranial arterial anomalies in 70 patients with PHACE syndrome. AJNR Am J Neuroradiol. 2010;31(10):1980–1986.
24. Girard C, Bigorre M, Guillot B, Bessis D. PELVIS Syndrome. Arch Dermatol. 2006;142(7):884–888.
25. Stockman A, Boralevi F, Taïeb A, Léauté-Labrèze C. SACRAL syndrome: spinal dysraphism, anogenital, cutaneous, renal and urologic anomalies,
associated with an angioma of lumbosacral localization. Dermatology. 2007;214(1):40–45.
26. Glick ZR, Frieden IJ, Garzon MC, Mully TW, Drolet BA. Diffuse neonatal hemangiomatosis: an evidence-based review of case reports in the
literature. J Am Acad Dermatol. 2012;67(5):898–903.
27. Yeh I, Bruckner AL, Sanchez R, Jeng MR, Newell BD, Frieden IJ. Diffuse infantile hepatic hemangiomas: a report of four cases successfully managed
with medical therapy. Pediatr Dermatol. 2011;28(3):267–275.
28. Berenguer B, Mulliken JB, Enjolras O, Bellini C, Burrows PE, Sundel RP, et al Rapidly involuting congenital hemangioma: clinical and histopathologic features. Pediatr Dev Pathol. 2003;6(6):495–510.
29. Mulliken JB, Enjolras O. Congenital hemangiomas and infantile hemangioma: missing links. J Am Acad Dermatol. 2004;50(6):875–882.
30. Patrice SJ, Wiss K, Mulliken JB. Pyogenic granuloma (lobular capillary hemangioma): a clinicopathologic study of 178
cases. Pediatr Dermatol. 1991;8(4):267–276.
31. Harris MN, Desai R, Chuang TY, Hood AF, Mirowski GW. Lobular capillary hemangiomas: An epidemiologic report, with emphasis on cutaneous
lesions. J Am Acad Dermatol. 2000;42(6):1012–1016.
32. Kroumpouzos G, Cohen LM. Dermatoses of pregnancy. J Am Acad Dermatol. 2001;45(1):1–19; quiz 19–22.
33. Eshkevari S, Kabir S, Alizadeh N, Nickhaha N. Curettage and punch excision of the vascular base: an effective method for treatment
of pyogenic granuloma with excellent results. Iran J Dermatol. 2014;17(69):91–5.
34. Croteau SE, Gupta D. The clinical spectrum of kaposiform hemangioendothelioma and tufted angioma. Semin Cutan Med Surg. 2016;35(3):147–152.
35. Kelly M. Kasabach-Merritt phenomenon. Pediatr Clin North Am. 2010;57(5):1085–1089.
36. McDaniel CG, Adams DM, Steele KE, Hammill AM, Merrow AC, Crane JL, et al Kaposiform lymphangiomatosis: Diagnosis, pathogenesis, and treatment. Pediatr Blood Cancer. 2023;70(4):e30219.
37. Carqueja IM, Sousa J, Mansilha A. Vascular malformations: classification, diagnosis and treatment. Int Angiol. 2018;37(2):127–142.
38. Kanada KN, Merin MR, Munden A, Friedlander SF. A prospective study of cutaneous findings in newborns in the United States: correlation
with race, ethnicity, and gestational status using updated classification and nomenclature. J Pediatr. 2012;161(2):240–245.
39. Dutkiewicz AS, Ezzedine K, Mazereeuw-Hautier J, Lacour JP, Barbarot S, Vabres P, et al A prospective study of risk for Sturge-Weber syndrome in children with upper facial
port-wine stain. J Am Acad Dermatol. 2015;72(3):473–480.
40. Roach ES. Neurocutaneous syndromes. Pediatr Clin North Am. 1992;39(4):591–620.
41. Adams ME, Aylett SE, Squier W, Chong W. A spectrum of unusual neuroimaging findings in patients with suspected Sturge-Weber
syndrome. AJNR Am J Neuroradiol. 2009;30(2):276–281.
42. Gonzalez CD, Mcdonald J, Stevenson DA, Whitehead KJ, Petersen MG, Presson AP, et al Epistaxis in children and adolescents with hereditary hemorrhagic telangiectasia. Laryngoscope. 2018;128(7):1714–1719.
43. Faurschou A, Olesen AB, Leonardi-Bee J, Haedersdal M. Lasers or light sources for treating port-wine stains. Cochrane Database Syst Rev. 2011;(11):CD007152.
44. Marqués L, Núñez-Córdoba JM, Aguado L, Pretel M, Boixeda P, Nagore E, et al Topical rapamycin combined with pulsed dye laser in the treatment of capillary vascular
malformations in Sturge-Weber syndrome: phase II, randomized, double-blind, intraindividual
placebo-controlled clinical trial. J Am Acad Dermatol. 2015;72(1):151–8.e1.
45. Longstreet B, Balakrishnan K, Saltzman B, Perkins JA, Dighe M. Prognostic value of a simplified anatomically based nomenclature for fetal nuchal
lymphatic anomalies. Otolaryngol Head Neck Surg. 2015;152(2):342–347.
46. Cash H, Bly R, Masco V, Shwayhat EU, Lutz DR, Van Tine LP, et al Prenatal Imaging Findings Predict Obstructive Fetal Airways Requiring EXIT. Laryngoscope. 2021;131(4):E1357–E1362.
47. Dighe MK, Peterson SE, Dubinsky TJ, Perkins J, Cheng E. EXIT procedure: technique and indications with prenatal imaging parameters for assessment
of airway patency. Radiographics. 2011;31(2):511–526.
48. de Serres LM, Sie KC, Richardson MA. Lymphatic malformations of the head and neck. A proposal for staging. Arch Otolaryngol Head Neck Surg. 1995;121(5):577–582.
49. Bonilla-Velez J, Whitlock KB, Ganti S, Theeuwen HA, Manning S, Bly R, et al Active Observation as an Alternative to Invasive Treatments for Pediatric Head and
Neck Lymphatic Malformations. Laryngoscope. 2021;131(6):1392–1397.
50. Luks VL, Kamitaki N, Vivero MP, Wang K, Ameen N, Bader A, et al Lymphatic and other vascular malformative/overgrowth disorders are caused by somatic
mutations in PIK3CA. J Pediatr. 2015;166(4):1048–54.e1–5.
51. Zenner K, Cheng CV, Jensen DM, Timms A, Shivaram G, Bly R, et al Genotype correlates with clinical severity in PIK3CA-associated lymphatic malformations. JCI Insight. 2019;4(21).
52. Keppler-Noreuil KM, Rios JJ, Parker VER, Semple RK, Lindhurst MJ, Sapp JC, et al. PIK3CA-related overgrowth spectrum (PROS): diagnostic and testing eligibility criteria,
differential diagnosis, and evaluation. Am J Med Genet A. 2015;167A(2):287–295.
53. Kurek KC, Luks VL, Ayturk UM, Alomari AI, Fishman SJ, Spencer SA, et al Somatic mosaic activating mutations in PIK3CA cause CLOVES syndrome. Am J Hum Genet. 2012;90(6):1108–1115.
54. Balakrishnan K, Menezes MD, Chen BS, Magit AE, Perkins JA. Primary surgery vs primary sclerotherapy for head and neck lymphatic malformations. JAMA Otolaryngol Head Neck Surg. 2014;140(1):41–45.
55. Bonilla-Velez J, Whitlock KB, Ganti S, Theeuwen HA, Manning SC, Bly RA, et al Acetylsalicylic acid suppression of the PI3K pathway as a novel medical therapy for
head and neck lymphatic malformations. Int J Pediatr Otorhinolaryngol. 2021;151:110869.
56. Fraulin FO, Flannigan RK, Sharma VK, McPhalen DF, Harrop RA. The epidemiological profile of the Vascular Birthmark Clinic at the Alberta Children's
Hospital. Can J Plast Surg. 2012;20(2):67–70.
57. Hung JWS, Leung MWY, Liu CSW, Choi CHT, Wong KW, Kwan HN. Venous Malformation and Localized Intravascular Coagulopathy in Children. Eur J Pediatr Surg. 2017;27(2):181–184.
58. Koo KSH, Dowd CF, Mathes EF, Sidhu K, Fuchs HE, Peters SM, et al MRI phenotypes of localized intravascular coagulopathy in venous malformations. Pediatr Radiol. 2015;45(11):1690–1695.
59. Dompmartin A, Ballieux F, Thibon P, Veyrac C, Verlaine M, Lacour JP, et al Elevated D-dimer level in the differential diagnosis of venous malformations. Arch Dermatol. 2009;145(11):1239–1244.
60. Soblet J, Limaye N, Uebelhoer M, Boon LM, Vikkula M. Variable Somatic TIE2 Mutations in Half of Sporadic Venous Malformations. Mol Syndromol. 2013;4(4):179–183.
61. Limaye N, Wouters V, Uebelhoer M, Pappas P, Mahoney W, Van Agtmael T, et al Somatic mutations in angiopoietin receptor gene TEK cause solitary and multiple sporadic
venous malformations. Nat Genet. 2009;41(1):118–124.
62. Eerola I, Boon LM, Mulliken JB, Aylett A, Dompmartin A, Lemaire A, et al Capillary malformation-arteriovenous malformation, a new clinical and genetic disorder
caused by RASA1 mutations. Am J Hum Genet. 2003;73(6):1240–1249.
63. Wouters V, Limaye N, Uebelhoer M, Ghosh S, Kim D, D'Amore PA, et al Hereditary cutaneomucosal venous malformations are caused by TIE2 mutations with widely
variable hyper-phosphorylating effects. Eur J Hum Genet. 2010;18(4):414–420.
64. Brouillard P, Boon LM, Revencu N, McCuaig C, Vikkula M, Pannett A, et al Genotypes and phenotypes of 162 families with a glomulin mutation. Mol Syndromol. 2013;4(4):157–164.
65. Soblet J, Kangas J, Nätynki M, Järvinen P, Eerola I, Vikkula M, et al Blue Rubber Bleb Nevus (BRBN) Syndrome Is Caused by Somatic TEK (TIE2) Mutations. J Invest Dermatol. 2017;137(1):207–216.
66. Seront E, Boon LM, Vikkula M. TEK-Related Venous Malformations. In: Adam MP, Feldman J, Mirzaa GM, et al, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2024. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1967/. Last updated Mar 2, 2023; cited Oct 16, 2024.
67. Heit JJ, Do HM, Prestigiacomo CJ, McCuaig C, Wu Y, Kuo A, et al Guidelines and parameters: percutaneous sclerotherapy for the treatment of head and
neck venous and lymphatic malformations. J Neurointerv Surg. 2017;9(6):611–617.
68. Tieu DD, Ghodke B V, Vo NJ, Perkins JA. Single-stage excision of localized head and neck venous malformations using preoperative
glue embolization. Otolaryngol Head Neck Surg. 2013;148(4):678–684.
69. Liu AS, Mulliken JB, Zurakowski D, Fishman SJ, Greene AK. Extracranial arteriovenous malformations: natural progression and recurrence after
treatment. Plast Reconstr Surg. 2010;125(4):1185–1194.
70. Spetzler RF, Martin NA. A proposed grading system for arteriovenous malformations. J Neurosurg. 1986;65(4):476–483.
71. Al-Olabi L, Polubothu S, Dowsett K, Kwan H, Larkin J, Lee D, et al Mosaic RAS/MAPK variants cause sporadic vascular malformations which respond to targeted
therapy. J Clin Invest. 2018;128(4):1496–1508.
72. Nikolaev SI, Vetiska S, Bonilla X, Pannett A, Tsang R, Smith A, et al Somatic Activating KRAS Mutations in Arteriovenous Malformations of the Brain. N Engl J Med. 2018;378(3):250–261.
73. Couto JA, Huang AY, Konczyk DJ, DiGuglielmo C, Chang K, Wang C, et al Somatic MAP2K1 Mutations Are Associated with Extracranial Arteriovenous Malformation. Am J Hum Genet. 2017;100(3):546–554.
74. Revencu N, Boon LM, Mendola A, MacKenzie R, Ghosh S, Vikkula M, et al RASA1 mutations and associated phenotypes in 68 families with capillary malformation-arteriovenous
malformation. Hum Mutat. 2013;34(12):1632–1641.
75. Parambil JG. Hereditary Hemorrhagic Telangiectasia. Clin Chest Med. 2016;37(3):513–521.
76. Lekwuttikarn R, Lim YH, Admani S, Choate KA, Teng JMC. Genotype-Guided Medical Treatment of an Arteriovenous Malformation in a Child. JAMA Dermatol. 2019;155(2):256–257.
77. Hashimoto T, Matsumoto MM, Li JF, Lawton MT, Young WL, University of California SFBSG. Suppression of MMP-9 by doxycycline in brain arteriovenous malformations. BMC Neurol. 2005;5(1):1.
78. Elsenousi A, Aletich VA, Alaraj A. Neurological outcomes and cure rates of embolization of brain arteriovenous malformations
with n-butyl cyanoacrylate or Onyx: a meta-analysis. J Neurointerv Surg. 2016;8(3):265–272.
79. Kim JY, Kim DI, Do YS, Lee DY, Kim JH, Cho YP, et al Surgical treatment for congenital arteriovenous malformation: 10 years' experience. Eur J Vasc Endovasc Surg. 2006;32(1):101–106.
80. Cox JA, Bartlett E, Lee EI. Vascular malformations: a review. Semin Plast Surg. 2014;28(2):58–63.
81. Ng AT, Tower RL, Drolet BA. Targeted treatment of vascular anomalies. Int J Womens Dermatol. 2021;7(5 Part A):636–639.
82. Horii KA, Drolet BA, Frieden IJ, Munden A, Blei F, McCuaig C, et al Prospective study of the frequency of hepatic hemangiomas in infants with multiple
cutaneous infantile hemangiomas. Pediatr Dermatol. 2011;28(3):245–253.
83. Metry DW, Haggstrom AN, Drolet BA, Frieden IJ, Lammer EJ, Schaffer R, et al A prospective study of PHACE syndrome in infantile hemangiomas: demographic features,
clinical findings, and complications. Am J Med Genet A. 2006;140(9):975–986.
84. Garzon MC, Epstein LG, Heyer GL, Grieden IJ, Haggstrom AN, Drolet BA, et al PHACE Syndrome: Consensus-Derived Diagnosis and Care Recommendations. J Pediatr. 2016;178:24–33.e2.
85. Orlow SJ, Isakoff MS, Blei F. Increased risk of symptomatic hemangiomas of the airway in association with cutaneous
hemangiomas in a “beard” distribution. J Pediatr. 1997;131(4):643–646.
86. Weber FC, Greene AK, Adams DM, Blei F, Drolet BA, Leach JL, et al Role of imaging in the diagnosis of parotid infantile hemangiomas. Int J Pediatr Otorhinolaryngol. 2017;102:61–66.
87. Bly RA, Holdefer RN, Slimp J, Kelly S, Craig J, Stal S, et al Preoperative Facial Nerve Mapping to Plan and Guide Pediatric Facial Vascular Anomaly
Resection. JAMA Otolaryngol Head Neck Surg. 2018;144(5):418–426.
Caleb M. Allred, 1 B.A., Fourth year medical student. University of Washington, Seattle, WA, USA.
Kaitlyn B. Zenner, 2 M.D., Department of Pediatric Otolaryngology, Cincinnati Children's Hospital, Cincinnati,
OH, USA.
Juliana Bonilla-Velez, 3 M.D., Division of Pediatric Otolaryngology, Seattle Children's Hospital, Seattle,
WA, USA; University of Washington School of Medicine; Department of Otolaryngology-Head
and Neck Surgery, University of Washington School of Medicine; Center for Clinical
and Translational Research, Seattle Children's Research Institute, Seattle, WA, USA.
About the Author: Caleb Allred received his bachelor's degree from Brigham Young University-Idaho in
Biomedical Sciences and received a minor in business management. He is a third-year
medical student at The University of Washington School of Medicine and has received
various awards for his work related to pediatric otolaryngology including the inaugural
Health Equity in Research Award from The American Society for Pediatric Otolaryngology
and the Best Research Presentation (1st and 2nd place) and Best Abstract for Original
Research (1st place) awards from the IJMS World Conference of Medical Student Research.
Correspondence: Caleb M. Allred. Address: 1410 NE Campus Pkwy, Seattle, WA 98195, USA. Email: cmallred@uw.edu
Editor: Francisco J. Bonilla-Escobar;
Student Editors: Mauricio Saldaña Ruiz, Prabhav Tekam;
Proofreader: Laeeqa Manji;
Layout Editor: Julian A. Zapata-RiosSubmission: Apr 22, 2024;
Process: Peer-reviewed
Cite as
Allred CM, Zenner KB, Bonilla-Velez J. Vascular Anomalies Review of the Head and Neck
for Physicians in Training. Int J Med Stud. 2024 Jul-Sep;12(3):284-293.
Copyright © 2024 Caleb M. Allred, Kaitlyn B. Zenner, Juliana Bonilla-Velez
This work is licensed under a Creative Commons Attribution 4.0 International License.
International Journal of Medical Students, VOLUME 12, NUMBER 3, September 2024