Letter to the Editor
Is it all in our Heads? The Role of CaMKII in Neurogenic Hypertension
Nathaniel Edward Hayward1, Paul MacDaragh Ryan1, Ryan Taylor Sless1
doi: http://dx.doi.org/10.5195/ijms.2018.272
Volume 6, Number 3: 129-131
Received 13 06 2018:
Accepted 20 09 2018
The letter
We wish to draw the attention of your readership to an intriguing development in neurogenic
hypertension that was reported in the journal of Neuroscience late last year.1 While 1 in every 3 American adults now experience elevated blood pressures, the majority
present with primary hypertension, the pathology of which is incompletely understood.
It has been postulated that many cases of treatment refractory primary hypertension
may be of a neurological origin 2 – i.e. sympathetically-driven increases in the vasoconstrictor tone of resistance vessels,
leading to elevated arterial blood pressure.3
The presympathetic neurons of the hypothalamic paraventricular nucleus (PVN) regulate
sympathetic outflow through projections to the rostral ventrolateral medulla. It is
therefore biologically plausible that hyperactivity of this pathway may contribute
towards hypertension.4 Increases in glutamatergic output on N-methyl-D-aspartate receptors (NMDARs) in the
PVN have previously been shown to increase vasomotor tone in a hypertensive rat model,
with increases in both presynaptic and postsynaptic NMDAR activity in PVN neurons.4 These NMDARs are activated through phosphorylation by numerous kinases including
the calcium/calmodulin-dependent protein kinase II (CaMKII), which itself is activated
by increases in cytoplasmic calcium.3 Although there is a strong association between CaMKII and NMDA activity, its role
in the hypertension-promoting PVN NMDAR activity currently remains unclear. Therefore,
Li et al. set out to determine the role of CaMKII in regulating synaptic NMDAR activity of
PVN presympathetic neurons and sympathetic motor tone in spontaneously hypertensive
rats (SHRs).1
Elevated sympathetic outflow has previously been implicated in the development of
essential hypertension in SHRs. Sympathetic outflow is regulated via PVN projections
to the rostral ventrolateral medulla and the intermediolateral cell column of the
spinal cord, which were the targets of these experiments. The major strength of this
study lies in the meticulous confirmation of PVN location, which was predicated on
several previous proof-of-principle experiments.4-6 In the current study, Li and colleagues test the role of both pre- and postsynaptic
CaMKII modulation of NMDARs by selective blockade of the suspected constituents involved
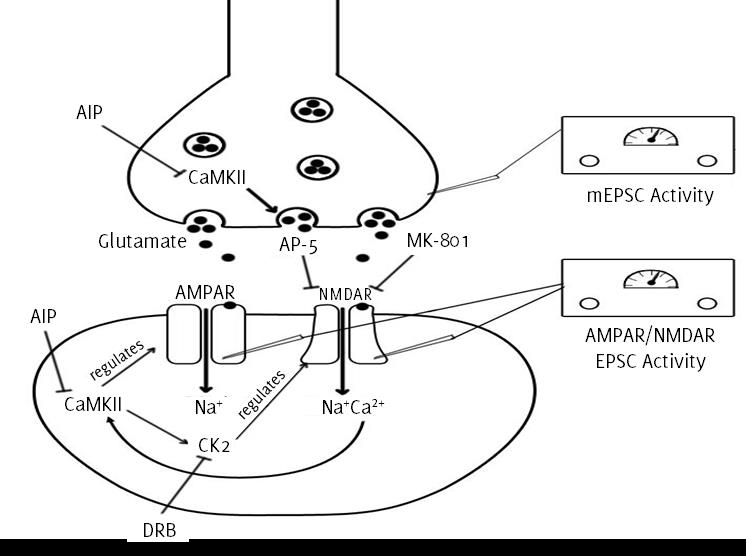
in elevated sympathetic outflow in SHRs, as summarised in Figure 1.
Figure 1.
Summary of pathways in the PVN of the hypothalamus illustrating the pre- and postsynaptic
role of CaMKII and associated locations of EPSC measurement. The drugs used in the
study and their associated targets are also shown: AIP, autocamtide 2-related inhibitory
peptide; AP-5, 2-amino-5-phosphonopentanoic acid; DRB, 5,6-Dichloro-1-β-D-ribofuranosylbenzimidazol;,
MK-801, Dizocilpine; CaMKII, Calcium/Calmodulin dependent protein kinase II; CK2,
Casein kinase 2; AM PAR, AMPA receptor; NMDAR, NMDA receptor; EPSC, Excitatory post-synaptic
current; mEPSC, miniature excitatory post-synaptic current.
Coronal brain slices were incubated in autocamtide 2-related inhibitory peptide (AIP),
a selective CaMKII inhibitor, and electrophysiological recordings of the hypothalamus
were made. CaMKII blockade normalized both the inherent raised baseline amplitude
of NMDAR-excitatory postsynaptic current (EPSC) and the NMDAR-EPSC/AMPAR-EPSC ratio
in SHRs compared to the control group. Subsequent puff-application of NMDA on post-synaptic
NMDARs was shown to increase SHR receptor current, while receptor currents were not
increased in Wiskar-Kyoto control rats suggesting the role of PVN NMDAR activity in
the pathogenesis of spontaneous hypertension. AIP blockade in conjunction with puff
NMDA diminished SHR receptor current, indicating the direct role of CaMKII on increased
postsynaptic activity in SHRs.
In attempt to assess the specific presynaptic role of CaMKII, miniature-EPSC (mEPSC)
activity was measured with NMDAR channels blocked by Dizocilpine (MK-801), a non-competitive
NMDAR antagonist. The blockade significantly increased mEPSC frequency in SHRs, which
was subsequently normalized by application of 2-amino-5-phosphonopentanoic acid (AP-5),
a competitive NMDAR antagonist. Increased mEPSC can thus be directly attributed to
NMDAR activity. To illustrate the role of CaMKII in this pathway, slices were incubated
once again with AIP and a decreased mEPSC frequency was observed. Subsequent application
of AP-5 had no effect suggesting that CaMKII is directly responsible for the tonic
basal increase in SHR PVN activity.
We currently know that expression of NMDARs is modulated by casein kinase (CK)2-mediated
phosphorylation of receptor subunits, which is regulated by CaMKII. Li et. al. previously demonstrated the role of 5,6-Dichloro-i-β-D-ribofuranosylbenzimidazole
(DRB), a selective CK2 inhibitor, in reducing mEPSC activity in the PVN of SHRs.3 However, AIP treatment coupled with DRB was not shown to further decrease either
NMDAR current amplitude or mEPSC frequency versus AIP treatment alone. This indicates
a common role of CaMKII and CK2 in both pre- and postsynaptic presympathetic neurons
of SHRs.
Intriguingly, Li et al. revealed raised CaMKII phosphorylation of the CIUN2M subunit exclusive to the PVN
of SHRs versus controls through Western blot analysis,1 while celiac ganglionectomy surgery did not reduce CaMKII phosphorylation levels
compared to sham surgery. This indicates that high blood pressure does not directly
increase CaMKII phosphorylation. Importantly, the authors further probed this result
by injecting AIP directly into the PVN. In turn, results observed demonstrated a reduction
in lumbar sympathetic nerve activity as well as arterial blood pressure in SHR.1 A similar effect was found with exclusive AP-5 injection, which suggests that CaMKII
is responsible for the increased sympathetic activity in SHRs.
The authors acknowledge that it is currently unclear as to the role of NMDAR activity
of hypothalamic PVN presympathetic neurons in secondary hypertensive states, such
as salt and obesity-induced hypertension. In this regard, a porcine model of mineralocorticoid-induced,
metabolic syndrome-associated hypertension may represent a useful tool in exploring
the potential role of this pathway.’ Overall the study was well designed as the involved
pathways were isolated with utmost precision with a focus on probing potential redundancies
in the CaMKII-mediated increase in vasomotor tone in SHRs. The stringent attention
to detail removed the effect of peripheral mediators in order to define the role of
CaMKII alone on the vasomotor pathway. Sequential blockade of constituents in both
the presynaptic and postsynaptic environment, aided in illustrating the direct role
of CaMKII in raised sympathetic outflow.
This data represents a significant step in our understandings of neurogenic hypertension
molecular underpinnings and provides novel information regarding the central role
of CaMKII in synaptic plasticity, thereby revealing potential targets for the future
development of pharmacologic treatments. However, further in vivo models are warranted to quantify the degree of antihypertensive effects prior to
conclusively targeting this pathway for the treatment of hypertension. Previously,
there had been indications that treatment-resistant, neurological pathology was mediated
by neuro-inflammation – more specifically, the chronic inflammation of the hypothalamic
PVN mediated by obesity and the renin angiotensin system.8 Indeed, it is difficult to determine whether this represents an entirely alternate
hypothesis of neurogenic hypertension or, rather, a contributory/resultant factor
of the neuropathology.
While this research is primarily an academic advancement for the field, it is difficult
not to extrapolate the translational potential. Despite the armoury of pharmaceuticals
currently available, essential hypertension is managed satisfactorily in less than
half of patients.9 Indeed, it has been proposed that much of this discrepancy may be neurologic in origin,10 indicating that the CaMKII pathway may represent a novel molecular target in the
fight against hypertension. We must now assess whether there are any suitably selective
drugs currently available, such as the NMDA receptor antagonist Memantine or whether
a novel therapy could be designed for this purpose. In terms of the alternate PVN
inflammation hypothesis, currently available pharmaceuticals such as angiotensin receptor
blockers, immunosuppressants and reactive oxygen species scavengers may offer potency
as adjunct therapeutics.
With such insightful data, it may now be time to ask ourselves: is hypertension all
in our heads?
Acknowledgments
None.
Conflict of Interest Statement & Funding
The Authors have no funding, financial relationships or conflicts of interest to disclose.
Conceptualization: NEH, PMR and RTS. Methodology: NEH, PMR and RTS. Investigation:
NEH, PMR and RTS. Writing – Original Draft: NEH, PMR and RTS. Writing – Review & Editing:
NEH, PMR and RTS. Visualization: NEH, PMR and RTS.
References
1.Li DP, Zhou JJ, Zhang J, Pan HL. CaMKII Regulates Synaptic NMDA Receptor Activity of Hypothalamic Presympathetic Neurons
and Sympathetic Outflow in Hypertension. J Neurosci. 2017 Nov 1;37(44):10690–9.
2.Mann SJ. Neurogenic essential hypertension revisited: the case for increased clinical and research
attention. Am J Hypertens. 2003 Oct;16(10):881–8.
3.Stocker SD, Kinsman BJ, Sved AF. Recent Advances in Neurogenic Hypertension: Dietary Salt, Obesity, and Inflammation. Hypertension. 2017 Jul 24. pii: hypertensionaha.117.08936.
4.Li DP, Pan HL. Role of gamma-aminobutytic acid (CABA)A and CABAB receptors in paraventricular nucleus
in control of sympathetic vasomotor tone in hypertension. J Pharmacol Exp Ther. 2007 Feb;320(2):615–26.
5.Ye ZY, Li DP, Li L, Pan HL. Protein Kinase CK2 Increases Glutamatergic Input in the Hypothalamus and Sympathetic
Vasomotor Tone in Hypertension. J Neurosci. 2011 Jun 1;31(22):8271–9.
6.Li DP, Zhu LH, Pachuau J, Lee HA, Pan HL. mCluRs Upregulation Increases Excitability of Hypothalamic Presympathetic Neurons
through NMDA Receptor Trafficking in Spontaneously Hypertensive Rats. J Neurosci. 2014 Mar 19:34(12):4309–17.
7.Schwarzl M, Hamdani N, Seiler S, Alogna A, Manninger M, Reilly S, . A porcine model of hypertensive cardiomyopathy: implications for heart failure with
preserved ejection fraction. Am J Physiol Heart Circ Physiol. 2015 Nov;309(9):H1407–18.
8.Winklewski PJ, Radkowski M, Wszedybyl-Winklewska M, Demkow U. Brain inflammation and hypertension: the chicken or the egg? J Neuroinflammation. 2015 May 3:12:85.
9.Materson BJ, Reda DJ, Cushman WC, Massie BM, Freis ED, Kochar MS, . Single-Drug Therapy for Hypertension in Men. A Comparison of Six Antihypertensive
Agents with Placebo. The Department of Veterans Affairs Cooperative Study Croup on
Antihypertensive Agents. N Engl J Med. 1993 Apr 1:328(13)1914–21.
10.Dudenbostel T, Siddiqui M, Charpure N, Calhoun DA. Refractory versus resistant hypertension: Novel distinctive phenotypes. J Nat Sci. 2017 Sep;3(9):e430.
Nathaniel Edward Hayward, 1 College of Medicine and Health, University College Cork, Cork, Ireland
Paul MacDaragh Ryan, 1 College of Medicine and Health, University College Cork, Cork, Ireland
Ryan Taylor Sless, 1 College of Medicine and Health, University College Cork, Cork, Ireland
Mihnea-Alexandru Găman, Editor
About the Author: Nathaniel Edward Hayward, M.Sc. is a student at the College of Medicine and Health,
University College Cork, Cork, Ireland.
Correspondence: Nathaniel Edward Hayward, M.Sc. Address: College Rd, University College, Cork, T12
K8AF, Ireland. Email: nathaniel.hayward@gmail.com
Cite as: Hayward NE, Ryan PM, Sless RT. Is it All in our Heads? The Role of CaMKII in Neurogenic Hypertension. Int J Med Students. 2018 Sep-Dec;6(3):129-131.
Copyright © 2018 Nathaniel Edward Hayward, Paul MacDaragh Ryan, Ryan Taylor Sless
This work is licensed under a Creative Commons Attribution 4.0 International License.
International Journal of Medical Students, VOLUME 6, NUMBER 3, December 2018