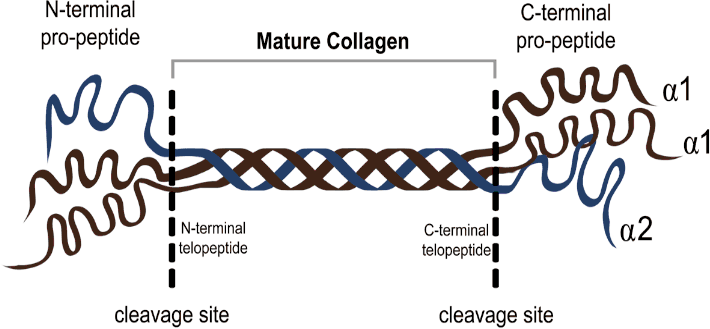
Formation of Mature Collagen from its Pro-Collagen Precursor, Releasing the Pro-Peptides at the N- and C-Terminals and in this Way Becoming more Stable and Resistant to Degradation.
Amerikos Argyriou1
doi: http://dx.doi.org/10.5195/ijms.2018.299
Volume 6, Number 3: 102-109
Received 08 08 2018: Accepted 14 12 2018
ABSTRACT
The Extracellular Matrix is a dynamic entity, showing constant degradation and deposition while providing the framework for the cardiomyocytes and interstitial proteins to lie on. Its function is important for the proper myocyte alignment within the heart and for internal communication from cell to matrix. Dysregulation of the remodeling process resulting in the breakdown of collagen by matrix metalloproteinases is a hallmark of heart failure pathophysiology and produces functional changes encompassing all matrix proteins. Several etiologies with distinct mechanisms ultimately bring about signs of heart exhaustion such as reduced ejection fraction, reduced compliance and ventricular dilatation. Discussed in this paper is the role of inflammation, collagen cross-linking and of myofibroblasts in matrix dysfunction and the mechanisms with which these changes occur in heart failure. Understanding extracellular protein roles within this context would allow for specific drug targeting and thus prevention of heart failure in the early stages of the disease. More studies must be conducted to discover the specific matrix proteins and cytokines that modulate the pathological remodeling process. Serum biomarkers of extracellular degradation products, selective metalloproteinase inhibitors and a personalized treatment approach with a révisai of the current classification of heart failure are topics requiring further exploration.
Keywords: Heart; Heart Failure; Extracellular Matrix; Cardiomyocytes; Matrix Metalloproteinases (Source: MeSH-NLM)..
Heart Failure is a global problem that is on the rise in the developed world. Worldwide, 38 million people are living with this disease, with a 50% increase within the last 15 years alone.1 With medical advances improving outcomes from myocardial infarctions (Ml) and congenital cardiac conditions, the same advances have also inadvertently brought about a steep increase in the prevalence of chronic heart failure.2 For the elderly in England and Wales prognosis can be worse than most cancers, with a 10% 30-day mortality following admission, highlighting the need to improve our understanding and treatment of this progressive disease.3 The extracellular matrix (ECM) is a scaffold including extracellular proteins, in which cardiac cells and cardiac vessels are arranged. We are now aware that the ECM is not a static structure but is actively broken down and reformed by an array of chemical and physical signaling within the heart.4 Changes in the myocardial matrix even encompass the cardiomyocytes, the resident cells of the myocardium that respond with a state of pathological hypertrophy.3 With heart failure, we see changes to the ECM that may differ in etiology, but all progressively results in an impaired contractility, fibrosis and eventual cavity dilatation.6 This review describes the ECM environment, identifies key modulators of its remodeling process in heart disease and finally mentions the clinical implications and future directions of this ever-growing field.
The ECM composition is vital for the heart’s structure as it regulates the alignment and activity of the cardiomyocytes, which are the resident cardiac contractility, which require a stable interstitium to properly function.’ Fibroblasts make up over 70% of cells in the myocardium and functionally secretes the most matrix proteins which is suggestive of the large quantities constantly being made in the interstitial space.4 The tight space between the myocytes must be able to transfer signals from the matrix to cell and also convert the individual contracting myofibrils into a collective pump, via the Collagen-lntegrin-Cytoskeleton-Myofibril relationship.8‘ The ECM includes structural, adhesive and regulatory proteins that work together to maintain the heart’s normal function.10 It is vital that all these proteins within the ECM stay balanced as they hold important roles in the maintenance of tissue homeostasis.
Fibrillar Collagen: In the heart, we can find collagen types I, III, IV, V, VI and VIII of which types I and III dominate the matrix. Type I provides tensile strength and type III distensibility.11-12 I, II, III, V and VI all have a common fibrillar structure and are not found on the basement membrane of cells.11 They all contain one main triple helix chain of Clycine-X-Y repeats where X is usually proline and Y can be any amino acid. The collagen helix terminates into 2 globular side chains on either side, referred to as the N- and C- Propeptides and these help in the alignment of the collagen strands as they are secreted from the cell.13 Upon maturation, the side-chains are cleaved by procollagen peptidases, releasing the Propeptides into the ECM and rendering the mature collagen insoluble.14 Cross-links between collagen fibrils are formed via the copper-containing enzyme, lysyl oxidase.13 This is a physiological occurrence that helps protect the collagen fibrils, both against the mechanical strains imposed on them as a result of the heart’s dynamic function, but also to enzymatic proteolysis resulting in degradation. Figure 1 shows the structure and cleavage site of a procollagen molecule. Collagen that has formed these cross-links is extremely stable and, pound for pound, is stronger than steel.16 The collagen in the ECM is constantly being remodeled under the control of fibroblasts that communicate with this scaffold via Discoidin Domain Receptor 2 (DDR2) surface receptors. These receptors allow collagen synthesis and degradation to be finely monitored by fibroblasts.17 The correct concentration of fibrillar collagen is vital for the heart’s form and function, and an inappropriate amount of deposition is associated with cardiovascular disease and more specifically diastolic heart failure.18
Figure 1.Formation of Mature Collagen from its Pro-Collagen Precursor, Releasing the Pro-Peptides at the N- and C-Terminals and in this Way Becoming more Stable and Resistant to Degradation.
Elastin: Elastin, unlike collagen, is not subject to a fixed turnover rate, and in a healthy myocardium can persist life-long.10 Elastin is implanted within its own scaffold of glycoprotein microfibrils once synthesized by fibroblasts, cardiomyocytes or endorhelial cells.20 The stiffness of the collagen fibers is counteracted upon by the elastic recoil properties of elastin, being up to a thousand times more flexible than collagen.9 This is what gives the heart the ability to rebound in diastole following a contraction. Elastin is also vulnerable to degradation in cardiac disease, either due to chronic stretch-recoil or due to increased protease activity of Matrix Metalloproteinases (MMP).21
These include all the non-collagenous proteins and account for over 95% of all the molecules in the ECM.22 They are vital for communication between cells and between the cell and the matrix.23 Yonggang et al studied their function in the setting of cardiac injury and concluded that they have a part to play in the response to injury, but individual roles and functions have yet to be identified.24 These proteins allow cardiomyocytes to monitor the myocardium and therefore modulate their behavior accordingly, through complex feedback loops.25
Collagen IV: This type of collagen is only found outside the membrane of cardiomyocytes where it folds into a sheet across the basal lamina.26 Collagen IV connects the contractile myofibril within the cardiomyocytes with the ECM and is therefore vital for the transfer of force across the cardiac tissue.27
Integrin: This cell-interstitium linking protein is made up of an α and β transmembrane subunit, as shown in Figure 2, and is required for nearly all communication within the ECM.28 Ross and Borg’s review on integrin role and function concludes that they are not merely adhesive proteins but have an important role in cell transduction in health and disease.29 Shed integrins have been found in heart failure by Ding et al, cleaved from distressed cardiomyocytes and dysregulating communication within the heart.30 Targeting this molecule for treatment has not been successful as there is complex sub-specialization within the integrin family where certain heterodimers bind fibronectin (α5β1) while others bind to collagen and laminin (α3β1 and α5β1).31
Figure 2.The Complex Cytoskeletal-Matrix Interaction as well as the Role of the Fibroblast in Contact with the Collagen Matrix via Discoidin Domain Receptor 2 Receptors and with the Myocite via Integrins.24,51,100 DDR2 is the Discoidin Domain Receptor 2.
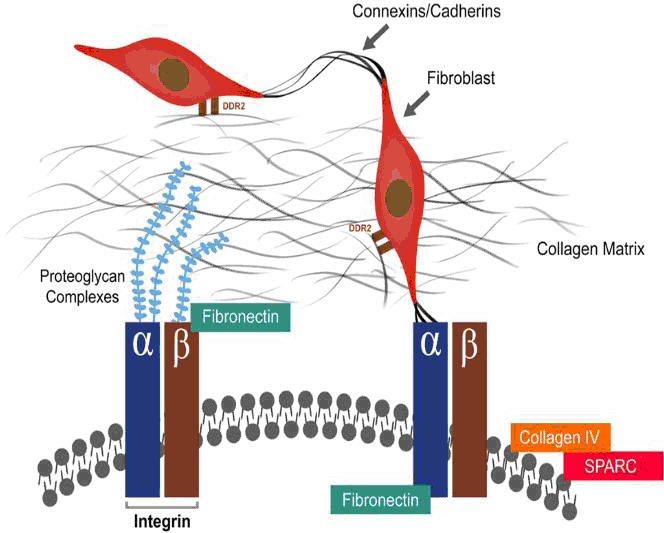
Fibronectin: This molecule exists as 3 different subtypes and can exist intracellularly and within the ECM (seen in Figure 2). It has been shown by Sharma et al to unfold itself and thus bind to integrin from within the cell’s basal lamina. In this way it assists in cell migration and differentiation. It is also produced following an injury by fibroblasts, macrophages and endorhelial cells to help link up the cells that form the scaffold in the injured area.32
SPARC (Osteonectin): SPARC stands for secreted protein acidic and rich in cysteine and is found in the basal lamina with many important functions. It stabilizes the collagen matrix by phosphorylation of the SMAD2 pathway with which it controls procollagen secretion out of the cell.33 It also facilitates cytokine and growth-hormone efficacy, regulates MMP expression and can alter cell shape.34-35 SPARC levels will markedly increase following cardiac injury, acute and chronic.24
One of the most noticeable changes to the ECM in the setting of heart failure is the dysregulation of collagen deposition and degradation. This fine control is performed by Matrix Metalloproteinases (MMPs) and Tissue Inhibitors of Metalloproteinases (TIMPs), two enzymes both formed within the fibroblast.36 The former is a protease enzyme and the latter its endogenous inhibitor with both being controlled by fibroblasts within the ECM. MMPs and TIMPs keep a homeostatic control over the collagenous matrix of the ECM as well as over the rest of the matrix proteins. As a result, the ECM is a dynamic entity constantly being broken down and reformed, in both health and disease. What separates a physiological state from a pathological one is the balance of the same remodeling process.
The matrix metalloproteinases are a family of over 24 zinc-containing endopeptidase enzymes. They are present in cardiovascular disease but can also be found in fibrosis and inflammation all over the body, as well as in tumor metastases.37 There are 6 main subtypes of MMP, and their enzymatic action dictates their title. They are all mentioned in Table 1 along with the substrate they degrade and the effects they produce. MMPs are released into the ECM as inactive zymogen molecules where they are activated by proteases such as plasmin or by another MMP.38-39 Spinale states that the catalytic domain is similar across all subtypes, but the extracellular binding domain, the C-terminal, is specific and conveys to each MMP its independent properties.40 In Table 1 are shown MMP-2 and MMP-9, the Celatinases, but can even break down Collagen types I, IV, V and the contractile apparatus from within a cell.41 Another MMP of importance in heart failure is MMP-14 or otherwise Membrane Type-i MMP (MTi-MMP). This class of protease, unlike the rest, exists only in an active form and can degrade all basement proteins as well as collagen.42 Tyagi as well as Nagase have both written an extensive list on the cytokines and hormones that they believe activate each MMP and have linked increased MMP activity to a heightened risk of heart failure (shown in Table 1). A factor that must be considered is the complex interaction between all the ECM components and how they are all intertwined. For example, it has been shown that MMP-3 might produce a shielding effect to disease, through an unclear downstream effect.43 Some MMPs, such as MMP-2, have shown pro-fibrotic capabilities and may actually cause a net increase in collage deposition rather than what we would expect, most likely viatheTGF-β pathway.44 To conclude, MMPs perform a diverse set of functions. On one hand they are able to degrade collagen but on the other hand also function to activate fibroblasts and increase collagen deposition. Radauceanu et al have discovered that ECM turnover is an independent predictor of all-cause mortality and heart failure hospitalization, therefore a very important aspect in the pathophysiology of Heart Failure.43 More studies into this field are necessary to more accurately elucidate individual MMP roles.
Table 1.Description of the Target and Effect of Each of the Main Matrix Metalloproteinases Implicated in Heart Failure.46,54,101-102
| MMP | Enzyme Family | Substrate | Result of Activation |
|---|---|---|---|
| MMP-l | Collagenase | C-Terminal Collagen 1 |
↑ Collagen breakdown |
| Basement Membrane Proteins | ↑ Myocardial Fibrosis | ||
| Pro-Inflammatory | |||
| Mesenchymal Cell Differentiation | |||
| MMP-2 | Gelatinase | Chondroitin Sulphate Proteoglycan | Vasoconstrictor |
| Angiostatin | Cell Apoptosis | ||
| Collagen III | ↑ TGF-β | ||
| ↑ Risk of Heart Failure | |||
| ↑ IGF-1 | |||
| MMP-3 | Stromelysin | Basement Membrane Proteins | Epithelial cell Apoptosis |
| Plasminogen | ↑ ICF-1 | ||
| E-cadherin | Pro-Inflammatory | ||
| Perlecan | Anti-Inflammatory | ||
| MMP-7 | Matrilysin | Fas Ligand | Adipocyte Differentiation |
| Rank Ligand | Fas-mediated Apoptosis | ||
| ECM proteins | Pro-Inflammatory | ||
| Precursor of TNF-a | Chemoattractant | ||
| MMP-9 | Gelatinase | Precursor of TGF-β | ↑ TGF-β |
| Citrate Synthase | ↓ IL-2 | ||
| ECM proteins | ↓ Mitochondrial Function | ||
| Collagen IV | Pro-Inflammatory | ||
| Plasminogen | ↑ Fibroblast migration | ||
| MMP-12 | Macrophage Elastase | ECM proteins | Pro-Inflammatory |
| Plasminogen | ↑ Fibroblast migration | ||
| Elastin | ↑ Endostatin-like fragment | ||
| MTi-MMP (MMP-14) | Stromelysin | ECM proteins | ↑ Myocardial Fibrosis |
| Collagen 1 | ↑ Risk of Heart Failure |
Legend: ↑ ICF-i means an increase in ICF-i as a result of that MMP being activated. ECM Proteins accounts for osteopontin, fibronectin, hyalutonan, tenascin-C and galectin-3 in these studies.
The TIMPs are folded disulphide-linked proteins that contain 2 domains: a C-terminal and an active N-terminal which binds to the active site of MMPs.46-47 This binding occurs due to cysteine residues at the N-terminal that react with the Zinc ion within the MMP active site.48 TIMP inhibition of MMPs has been shown to overpower MMP activity as certain studies of left ventricular hypertrophy and chronic heart failure where both MMP and TIMP levels were similarly increased resulted in fibrotic deposition and not degradation.44 Perhaps this is a marker of TIMP efficacy being superior in the balance of MMPs and TIMPS. TIMP-4 is believed to be a key player in terms of fibrotic deposition in the heart by Li et al, Schultz et al and Graham et al.49-51 Moore states that TIMP-2 and TIMP-3 are most abundant while TIMP-4 is not cardio-specific and only partly expressed by healthy cardiomyocytes.46 These minor discrepancies in the results could reflect the fact that more studies are required in order to understand the exact roles and populations of these protease inhibitors.
In a failing heart, many pathological events are occurring that have a large impact on the constituents of the heart’s tissues. Both the ECM and the cells of the heart are affected by an increase in inflammatory mediators and an increased dynamic load.32 We now know fibroblasts have the potential to signal pro-inflammatory mediators in the absence of infection or ischemia.32 This signaling occurs as a result of the heart’s inability to cope with mechanical demands and directly impacts the cardiac molecular environment.33 It is now well documented that ECM remodeling is a distinguished feature of cardiac risk that will gradually lead to heart failure.40 The key player we tend to focus on are the MMPs that have a direct functional role in the progression to heart failure by enzymatically breaking down the structure of the collagenous matrix.34 The question that remains is why do we see fibrotic deposition in some cases while in others we see collagen degradation? An interpretation of these seemingly conflicting phenotypes is in line with Kim et al’s transgenic mouse model. Mice given an active MMP-i gene initially responded with myocyte hypertrophy, myofibroblast formation and increased collagen synthesis at 6-weeks. This matches the initial state of hypertrophy and fibrosis seen in early stage heart failure.33 However, at 12-months the collagen content of the transgenic mice was far lower than the wild-type control mice.36 The proposed mechanism is one of adapting to the stressor, this case the collagenase MMP-i, by synthesizing more collagen to the point of exhaustion. At this point we begin to see a destruction of the matrix and weakening of the heart wall, markers that we also see in human patients suffering from heart failure.36 These matrix changes directly influence the heart’s mechanical functioning and lead to the gradual signs and symptoms we associate with cardiac failure.
As we know, the ECM is constantly active and subject to remodeling and as a result is exceptionally susceptible to any chemical or mechanical stressors that might affect it.37 Inflammation due to increased cardiac stress induces cytokines such as TGF-β, IL-1β, FGF, Angiotensin II and Endothelin-i, which upregulate MMP activity and increase the migration of myofibroblasts into the heart.22,58–59 Damage associated molecular patterns (DAMPs) released from distressed cells promote further inflammation and were shown to activate fibroblasts in vitro.60 Bradham et al looked at zymographic activity in failing canine cardiac tissue and discovered decreased MMP activity in tissue injected with a TNF-α blocker.61 This suggests that TNF-α and potentially other cytokines and hormones do interact with ECM turnover and can worsen the progression to heart failure by directly interfering with the MMP/TIMP control of the ECM. It should be stated that some attempts to tackle these mediators are being pursued in current specialized centres.62 This inflammatory response is not limited to collagen but also affects fibronectin, osteonectin, elastin and laminin, which were all found to be increased in heart failure pathology by Hein et al.63 Finally, inflammatory mediators can serve as transcription factor activators for MMPs, tipping the scale of collagen homeostasis by direct MMP upregulation.64
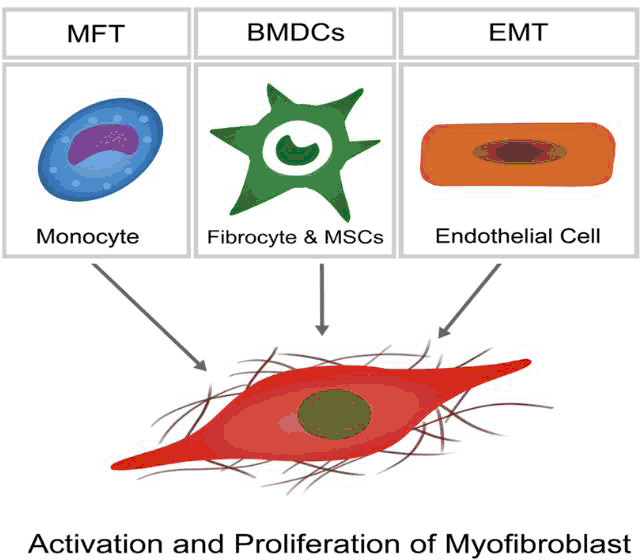
Myofibroblasts, which are commonly absent in healthy myocardium having been identified only in heart valve leaflets, are key drivers of ECM remodeling in disease (Progenitor populations shown in Figure 3)65-66. They perform all the functions of an active fibroblast to an even greater degree to cause and increase in matrix deposition and fibrosis.65 They also exhibit a myocyte-like phenotype with contractile capabilities and the generation of a stress-fiber network.67 The matrix deposited by these cells is also said to differ to that of fibroblast origin, with particularly increased amounts of Collagen I and Fibronectin.68 All these features lead to a change in the normal environment both structurally and composition wise in the setting of heart failure. In the acute setting myofibroblasts are generated from cardiac pericytes, fibroblasts and smooth muscle cells. Long-term stressor persistence allows myofibroblasts to form from cardiac endorhelial cells that transition into mesenchymal cells and also from cells derived from the hematopoietic stem cell line (Figure 3).69 Myofibroblasts contribute to pathological hypertrophy, usually with accompanied diastolic dysfunction.70 This dysfunction is a direct result of the fibrotic stiffness brought about by the myofibroblasts, as described above. Overall, the changes described result in a stiffer, less compliant myocardium that is less effective in its ability to pump blood around the body.71 Myofibroblasts damage the cardiac environment in heart failure but in an Ml can have beneficial properties and be necessary for wound healing.72-74 Interestingly, several animal studies have been pursued in the beneficial treatment using myofibroblast differentiation from progenitor populations such as BMDCs in the response to acute ischemic damage post-MI.75
Figure 3.The Different Progenitor Populations that Differentiate into Myofibroblasts in the Setting of Injury and Mediate further Remodeling.50,55,65.
Collagen Cross-linking (CCL) is a feature of ECM remodeling in heart failure16-76 and has been shown by Graham et al to be more indicative of a worsening prognosis than collagen type ratios in the ECM.37 CCL occurs via two different mechanisms in the heart: an enzyme-mediated pathway and a pathological proteoglycation pathway.37 The latter is most evident with aging and in diabetics, but can occur incidentally with other groups too.” The end result is an increased stiffness of the myocardium and ventricular dilatation, both increasing the risk of heart failure.78 As previously described, collagen must be cross-linked to increase its strength and resistance to degradation, and a collagen matrix with decreased CCL is a weaker and more unstable structure. Gunja-Smith et al investigated the amount of pyridinoline CCL in human heart collagen directly post cadaveric transplant and found that even though collagen levels were quadrupled in some specimen, the overall CCL between that collagen was poor, resulting in dilatation and decreased compliance, both clinical features of heart disease. 79 This study is suggestive of the fact that even though a fibrotic state exists in heart failure (study in question looked at dilated cardiomyopathy), the collagen of these patients although plentiful is weak as a result of its poor cross-linking.
In current specialized centers, it is standard practice for multiple cardiac biomarkers to be monitored in patients with heart failure. Several biomarkers including BNP, NT-pro BNP, Troponins as well as proteins like Galectin-3 are screened for to help in disease prognosis.80-81.
With the recent advances of cardiac matrix proteomics, ECM biomarkers may also be monitored. As previously described, collagen Propeptides are released into the ECM and are picked up in blood tests as a measure of collagen synthesis. Collagen telopeptides on the other hand should not be cleaved in the physiological state, therefore C- and N-telopeptides in serum can point at pathological collagen degradation.82 These biomarkers are good indicators of what is going on in the ECM and are found in our bloodstream before any physical symptom of heart failure is apparent. Using these biomarkers in a day-to-day clinic for cardiac observation should be explored further as the N-terminal propeptide of Type I and the C-terminal of Type III Collagen have been shown to statistically correlate with myocardial fibrosis.43-83-84 Furthermore, Kato et al highlighted the prognostic potential of using other ECM biomarkers in the clinic as osteopontin and MMP/TIMP ratios all decrease significantly post-LVAD (Left Ventricular Assist Device) implantation in patients with heart failure and were found to be consistently higher in those who suffered with right ventricular failure in the months following the surgery.83 A final reason to further explore this area of research, as opposed to looking at the ECM under a microscope, are the barriers with attaining live cardiac biopsies which although proven relatively safe in practice, having them performed for research purposes is not ethically justifiable.86 A Review written by Mr. R.J. van Kimmenade summarizes the novel biomarkers in Heart Failure.87
Since Weber et al linked the Renin-Angiotensin-Aldosterone-System (RAAS) with eventual myocardial fibrosis and stiffness in 1994 we have treated most forms of heart failure thus far with Diuretics, Anticoagulants, Digoxin, Beta-Blockers, ACE Inhibitors and Aldosterone Inhibitors.88-90 We have recently discovered that inflammatory markers inducing MMP activation are in play even before the RAAS system is activated in heart failure.39,91-92 Therefore, MMP inhibition could be considered as a potential early intervention strategy. Positive results depicting a decrease in collagen deposition have been recorded in animal trials yet the transfer to human trials has been slow due to the side-effects brought about by the many beneficial and vital functions of the MMPs.93 Selectively identifying an MMP causing pathological remodeling without a concomitant role in health has proven difficult with limited efficacy in the former and significant musculoskeletal side-effects in the latter.94 Nevertheless, the future of early treatment in patients with heart failure could very well lie in treating their specific matrix pathology, and not in purely treating the neurohormonal systemic symptoms, as is current practice.
Miner and Miller suggested that the future of heart failure medicine will be tailoring treatment to each patient’s specific condition and body.57 As Table 2 describes, there are many different routes to acquiring heart failure and they will all likely produce very different inflammatory responses to disease with differing amounts and type of collagen remodeling. As a result of relatively recent research, we now know that collagen consistency will widely differ amongst the population in the amount of cross-linked collagen fibers they will exhibit, affected by disease and age.23,44 In a canine trial, Jugdutt showed that age affected an animal’s response to heart failure medication post-MI.95 Also, as Table 3 will highlight, different causes of heart failure all vary in their exact mechanism and pathophysiology and therefore require specific and individual treatment.96 Following a one-fits-all treatment plan might not be the most successful approach considering the vast amount of research that has been done on the cardiac ECM until now.
Table 2.The Different Etiological Subtypes of Cardiomyopathy along with Their Mechanism, Biomarkers and Clinical Features.46,103-105
| Ischemic | Idiopathic | Hypertrophic | Hypertensive | Sources | |
|---|---|---|---|---|---|
| Mechanism | Fibroblast Differentiation into Myofibroblasts | Genetic mutation of collagen turnover (reported hyperactive MMP-1) | TGF-B driven fibrosis | RAAS driven perivascular fibrosis | Tyagi, 1998 |
| Inflammatory Cascade | |||||
| Biomarkers | ↑ MMP-1, ↑ MMP-2 | ↑ TGF-B, ↑ MMP-2 | ↑ TGF-B, ↑ Angiotensin ll/Aldosterone | Vilahur el al, 2012 | |
| ↓ TIMP-1, ↓ TIMP-3, ↓ TIMP-4 | ↓ TIMP-4 | ↑ MMP-2, ↑ TIMP-1, ↑ TIMP~2 | ↑ MMP-2 | Moore et al, 2011 | |
| Features | Dilatation in infarcted zone | Dilatation | Hypertrophy (matrix and myocytes) | ↑ Collagen deposition | Vilahur et al. 2012 |
| Hypertrophy in non-infarcted zone | Hypertrophy | Fibrosis | Myocyte Hypertrophy | Bradford, 2007 | |
| Collagen deposition | Systolic Failure | Diastolic Failure | Perivascular Collagen |
Legend: MMP- Matrix Metalloproteinase, TIMP-Tissue Inhibitor of Metalloproteinase.
Heart Failure is a complex and progressive disease involving both the cells and ECM of the myocardium. A tip in the fine balance of ECM dynamic remodeling, mostly concerning MMPs and TIMPs, affects the heart’s physiological properties immensely and can lead to the dysregulation that provides the basis for progression to heart failure in the future. These imbalances within the myocardium occur through distinct mechanisms of variable root cause, as shown in Table 2, with the final result remaining the breakdown of a stable matrix and the common pathophysiology that will ensue. The current research field requires further clarification into the key protein modulators that have an effect on the ECM in disease, other than the enzymatic MMPs and TIMPS. Although many discovered, it is yet unclear as to which changes are primary or secondary to the remodeling process. Zamilpa and Lindsey concluded with a similar view in the need for a catalogue on the specific changes that occur in the ECM with cardiac injury.97 Many studies have pointed out Osteopontin, Galectin, Periostin and Integrin as potential modulators.8,22,29,32,84,98-99 The near future will help us get an even clearer picture of the roles and importance of these non-collagenous proteins in heart failure. The field of the Cardiac ECM holds many academic and clinical applications and we have yet to realize its true potential in combatting the chronic issue of heart failure.
I acknowledge the advisory help received from Professor of Cardiac Pathophysiology Andrew Traffor (BVSc, CertVA, PhD, MRCVS). There was no funding or endorsement from any third party. All the work shown is truthful, objective and with no agenda on mine or my Supervisor's behalf.
No conflict of interest exists for this article. No financing was involved for this article.
Conceptualization: AA. Methodology: AA. Software: AA. Validation: AA. Formal Analysis: AA. Data Curation: AA. Investigation: AA. Resources: AA. Writing - Original Draft: AA. Writing - Review & Editing: AA. Visualization: AA. Supervision: AA. Project Administration: AA.
1.Braunwald E. The war against heart failure: the Lancet lecture. Lancet. 2015; Feb 28 385(9970): 812–24.
2.Smolina K, Wright FL, Rayner M, Coldacre MJ. Determinants of the decline in mortality from acute myocardial infarction in England between 2002 and 2010: linked national database study. BMJ. 2012; Jan 25 344: d8059.
3.Cleland JC, McDonagh T, Rigby AS, Yassin A, Whittaker T, Dargie HJ, . The national heart failure audit for England and Wales 2008-2009. Heart. 2011; Jun 97(11): 876–86.
4.Camelliti P, Borg TK, Kohl P. Structural and functional characterisation of cardiac fibroblasts. Cardiovasc Res. 2005; Jan 1 65(1): 40–51.
5.Meschiari CA, Ero OK, Pan H, Finkel T, Lindsey ML. The impact of aging on cardiac extracellular matrix. Ceroscience. 2017; Feb 39(1): 7–18.
6.Harris KM, Spirito P, Maron MS, Zenovich AC, Formisano F, Lesser JR, . Prevalence, clinical profile, and significance of left ventricular remodeling in the end-stage phase of hypertrophic cardiomyopathy. Circulation. 2006; JuI 18 114(3): 216–25.
7.Lindsey ML, Hall ME, Harmancey R, Ma Y. Adapting extracellular matrix proteomics for clinical studies on cardiac remodeling post-myocardial infarction. Clin Proteomics. 2016; 13: 19.
8.Eckhouse SR, Spinale FC. Changes in the myocardial interstitium and contribution to the progression of heart failure. Heart Fail Clin. 2012; Jan 8(1): 7–20.
9.Fomovsky CM, Thomopoulos S, Holmes JW. Contribution of extracellular matrix to the mechanical properties of the heart. J Mol Cell Cardiol. 2010; Mar 48(3): 490–6.
10.Parker KK, Ingber DE. Extracellular matrix, mechanotransduction and structural hierarchies in heart tissue engineering. Philos Trans R Soc Lond B Biol Sci. 2007; Aug 29 362(1484): 1267–79.
11.Burlew BS, Weber KT. Connective tissue and the heart. Functional significance and regulatory mechanisms. Cardiol Clin. 2000; Aug 18(3): 435–42.
12.Kitamura M, Shimizu M, Ino H, Okeie K, Yamaguchi M, Fujno N, . Collagen remodeling and cardiac dysfunction in patients with hypertrophic cardiomyopathy: the significance of type III and VI collagens. Clin Cardiol. 2001; Apr 24(4): 325–9.
13.Lodish HBA, Zipursky SL, . Molecular Cell Biology. 4th ed. New York: W.H. Freeman; 2000. Section 22.3: Collagen: The Fibrous Proteins of the Matrix p.
14.Ricard-Blum S, Ruggiero F. The collagen superfamily: from the extracellular matrix to the cell membrane. Pathol Biol (Paris). 2005; Sep 53(7): 430–42.
15.Holmes DF, Graham HK, Trotter JA, Kadler KE. STEM/TEM studies of collagen fibril assembly. Micron. 2001; Apr 32(3): 273–85.
16.Kato S, Spinale FC, Tanaka R, Johnson W, Cooper Ct, Zile MR. Inhibition of collagen cross-linking: effects on fibrillar collagen and ventricular diastolic function. Am J Physiol. 1995; Sep 269(3 Pt 2): H863–8.
17.Souders CA, Bowers SL, Baudino TA. Cardiac fibroblast: the renaissance cell. Circ Res. 2009; Dec 4 105(12): 1164–76.
18.McCurdy S, Baicu CF, Heymans S, Bradshaw AD. Cardiac extracellular matrix remodeling: fibrillar collagens and Secreted Protein Acidic and Rich in Cysteine (SPARC). J Mol Cell Cardiol. 2010; Mar 48(3): 544–9.
19.Vottelet M, Berrio DA, Hotke A, Sabatiet L, Reinhatdt DP, Nsair A, . Elastogenesis at the onset of human cardiac valve development. Development. 2013; Jun 140(11): 2345–53.
20.Mithieux SM, Weiss AS. Elastin. Adv Protein Chem. 2005; 70: 437–61.
21.Ashworth JL, Murphy C, Rock MJ, Sherratt MJ, Shapiro SD, Shuttleworth CA, . Fibrillin degradation by matrix metalloproteinases: implications for connective tissue remodelling. Biochem J. 1999; May 15 340 (Pt 1): 171–81.
22.Bowets SL, Banerjee I, Baudino TA. The extracellular matrix: at the center of it all. J Mol Cell Cardiol. 2010; Mar 48(3): 474–82.
23.Brower CL, Gardner JD, Forman MF, Murray DB, Voloshenyuk T, Levick SP, . The relationship between myocardial extracellular matrix remodeling and ventricular function. Eur J Cardiothorac Surg. 2006; Oct 30(4): 604–10.
24.Ma Y, Halade GV, Lindsey ML. Extracellular matrix and fibroblast communication following myocardial infarction. J Cardiovasc Transi Res. 2012; Dec 5(6): 848–57.
25.Parsons JT, Horwitz AR, Schwartz MA. Cell adhesion: integrating cytoskeletal dynamics and cellular tension. Nat Rev Mol Cell Biol. 2010; Sep 11(9): 633–43.
26.Hegarova M, Kautzner J. Changes in the extracellular matrix during myocardial remodelling. Curr Res Cardiol. 2015; 2: 35–9.
27.Bruggink AH, van Oosterhout MF, de Jonge N, Cleutjens JP, van Wichen DF, van Kuik J, . Type IV collagen degradation in the myocardial basement membrane after unloading of the failing heart by a left ventricular assist device. Lab Invest. 2007; Nov 87(11): 1125–37.
28.Shai SY, Hatpf AE, Ross RS. Integtins and the myocardium. Genet Eng. 2002:24:87–105.
29.Ross RS, Borg TK. Integrins and the myocardium. Cite Res. 2001 Jun; 88(11):1112–9.
30.Ding B, Price RL, Goldsmith EC, Borg TK, Yan X, Douglas PS, . Left ventricular hypertrophy in ascending aortic stenosis mice: anoikis and the progression to early failure. Circulation. 2000; Jun 20 101(24): 2854–62.
31.Hornberger LK, Singhroy S, Cavalle-Carrido T, Tsang W, Keeley F, Rabinovitch M. Synthesis of extracellular matrix and adhesion through beta(1) integrins are critical for fetal ventricular myocyte proliferation. Circ Res. 2000; Sep 15 87(6): 508–15.
32.Sharma A, Askari JA, Humphries MJ, Jones EY, Stuart DL. Crystal structure of a heparin- and integrin-binding segment of human fibronectin. EMBO J. 1999; Mar 15 18(6): 1468–79.
33.Rienks M, Papageorgiou AP, Frangogiannis NG, Heymans S. Myocardial extracellular matrix: an ever-changing and diverse entity. Circ Res. 2014; Feb 28 114(5): 872–88.
34.Bradshaw AD, Sage EH. SPARC, a matticellulat protein that functions in cellular differentiation and tissue response to injury. J Clin Invest. 2001; May 107(9): 1049–54.
35.de Castro Bras LE, Toba H, Baicu CF, Zile MR, Weintraub ST, Lindsey ML, . Age and SPARC change the extracellular matrix composition of the left ventricle. Biomed Res Int. 2014; 2014: 810562.
36.Lindsey ML, lyer RP, Jung M, DeLeon-Pennell KY, Ma Y. Matrix metalloproteinases as input and output signals for post-myocardial infarction remodeling. J Mol Cell Cardiol. 2016; Feb 91: 134–40.
37.Graham HK, Trafford AW. Spatial disruption and enhanced degradation of collagen with the transition from compensated ventricular hypertrophy to symptomatic congestive heart failure. Am J Physiol Heart Cite Physiol. 2007; Mar 292(3): Hi 364–72.
38.Tyagi SC, Kumar SC, Haas SJ, Reddy HK, Voelker DJ, Hayden MR, . Post-transctiptional regulation of extracellular matrix metalloproteinase in human heart end-stage failure secondary to ischemic cardiomyopathy. J Mol Cell Cardiol. 1996; Jul 28(7): 1415–28.
39.Lindsey ML. MMP induction and inhibition in myocardial infarction. Heart Fail Rev. 2004; Jan 9(1): 7–19.
40.Spinale FG. Matrix Metalloproteinases: Regulation and Dysregulation in the Failing Heart. Circulation Research. 2002; 90(5): 520–30.
41.Rouet-Benzineb P, Buhlet JM, Dteyfus P, Delcourt A, Dorent R, Perennec J, . Altered balance between matrix gelatinases (MMP-2 and MMP-9) and their tissue inhibitors in human dilated cardiomyopathy: potential role of MMP-9 in myosin-heavy chain degradation. Eur) Heart Fail. 1999; Dec 1(4): 337–52.
42.Hutchinson KR, Stewart JA, Jr., Lucchesi PA. Extracellular matrix remodeling during the progression of volume overload-induced heart failure. J Mol Cell Cardiol. 2010; Mar 48(3): 564–9.
43.Nagase H, Visse R, Murphy G. Structure and function of matrix metalloproteinases and TIMPs. Cardiovasc Res. 2006; Feb 15 69(3): 562–73.
44.Horn MA, Trafford AW. Aging and the cardiac collagen matrix: Novel mediators of fibrotic remodelling. J Mol Cell Cardiol. 2016; Apr 93: 175–85.
45.Radauceanu A, Ducki C, Virion JM, Rossignol P, Mallat Z, McMurray J, . Extracellular matrix turnover and inflammatory markers independently predict functional status and outcome in chronic heart failure. J Card Fail. 2008; Aug 14(6):467–74.
46.Moore L, Fan D, Basu R, Kandalam V, Kassiri Z. Tissue inhibitor of metalloproteinases (TIMPs) in heart failure. Heart Fail Rev. 2012; Sep 17(4-5): 693–706.
47.Jugdutt Bl. Remodeling of the myocardium and potential targets in the collagen degradation and synthesis pathways. Curt Drug Targets Cardiovasc Haematol Disord. 2003; Mar 3(1): 1–30.
48.Catetina NC, Windsot LJ, Bodden MK, Yermovsky AE, Taylor KB, Birkedal-Hansen H, . Clycosylation and NH2-terminal domain mutants of the tissue inhibitor of metalloproteinases-i (TIMP-i). Biochim Biophys Acta. 1998; Oct 14 1388(1): 21–34.
49.Li YY, Feldman AM, Sun Y, McTiernan CF. Differential expression of tissue inhibitors of metalloproteinases in the failing human heart. Circulation. 1998 Oct;98(17):1728–34.
50.Schultz CS, Wysocki A. Interactions between extracellular matrix and growth factors in wound healing. Wound Repair Regen. 2009; Mar-Apr 17(2): 153–62.
51.Graham HK, Horn M, Traffotd AW. Extracellular matrix profiles in the progtession to heart failure. European Young Physiologists Symposium Keynote Lecture-Bratislava 2007. Acta Physiol (Oxf). 2008; Sep 194(1): 3–21.
52.Suthahar N, Meijers WC, Sillje HHW, de Boer RA. From Inflammation to Fibrosis-Molecular and Cellular Mechanisms of Myocardial Tissue Remodelling and Perspectives on Differential Treatment Opportunities. Curr Heart Fail Rep. 2017; Aug 14(4): 235–50.
53.Westermann D, Lindner D, Kasner M, Zietsch C, Savvatis K, Escher F, . Cardiac inflammation contributes to changes in the extracellular matrix in patients with heart failure and normal ejection fraction. Circ Heart Fail. 2011; Jan 4(1): 44–52.
54.DeLeon-Pennell KY, Meschiati CA, Jung M, Lindsey ML. Matrix Metalloproteinases in Myocardial Infarction and Heart Failure. Prog Mol Biol Transi Sci. 2017: 147: 75–100.
55.Fan D, Takawale A, Lee J, Kassiri Z. Cardiac fibroblasts, fibrosis and extracellular matrix remodeling in heart disease. Fibrogenesis Tissue Repair. 2012; Sep 3 5(1): 15.
56.Kim HE, Dalai SS, Young E, Legato MJ, Weisfeldt ML, D’Armiento J. Disruption of the myocardial extracellular matrix leads to cardiac dysfunction. J Clin Invest. 2000: Oct 106(7): 857–66.
57.Miner EC, Millet WL. A look between the cardiomyocytes: the extracellular matrix in heart failure. Mayo Clin Proc. 2006: Jan 81(1): 71–6.
58.Manabe I, Shindo T, Nagai R. Gene expression in fibroblasts and fibrosis: involvement in cardiac hypertrophy. Circ Res. 2002; Dec 13 91(12): 1103–13.
59.Baum J, Duffy HS. Fibroblasts and myofibroblasts: what are we talking about? J Cardiovasc Phatmacol. 2011; Apr 57(4): 376–9.
60.Franssen C, Gonzalez Miqueo A. The role of titin and extracellular matrix remodelling in heart failure with ptesetved ejection fraction. Neth Heart J. 2016: Apr 24(4): 259–67.
61.Bradham WS, Moe G, Wendt KA, Scott AA, Konig A, Romanova M, . TNF-alpha and myocardial matrix metalloproteinases in heart failure: relationship to LV remodeling. Am J Physiol Heart Circ Physiol. 2002; Apr 282(4): H1288–95.
62.Oikonomou E, Tousoulis D, Siasos G, Zatomitidou M, Papavassiliou AG, Stefanadis C The role of inflammation in heart failure: new therapeutic approaches. Hellenic J Cardiol. 2011; Jan-Feb 52(1): 30–40.
63.Hein S, Schapet J. The extracellular matrix in normal and diseased myocardium. J Nucl Cardiol. 2001; Mar-Apr 8(2): 188–96.
64.Deschamps AM, Spinale FG. Pathways of matrix metalloproteinase induction in heart failure: bioactive molecules and transcriptional regulation. Cardiovasc Res. 2006; Feb 15 69(3): 666–76.
65.Kisseleva T, Brenner DA. Mechanisms of fibrogenesis. Exp Biol Med (Maywood). 2008; Feb 233(2): 109–22.
66.Sun Y, Weber KT. Infarct scat: a dynamic tissue. Cardiovasc Res. 2000; May 46(2): 250–6.
67.Pettov W, Fagatd RH, Lijnen PJ. Stimulation of collagen production by transforming growth factor-betai during differentiation of cardiac fibroblasts to myofibroblasts. Hypertension. 2002; Feb 39(2): 258–63.
68.Davis J, Molkentin JD. Myofibroblasts: trust your heart and let fate decide. J Mol Cell Cardiol. 2014; May 70: 9–18.
69.Hinz B. Formation and function of the myofibroblast during tissue repair. J Invest Dermatol. 2007; Mar 127(3): 526–37.
70.Burlew BS, Weber KT. Cardiac fibrosis as a cause of diastolic dysfunction. Herz. 2002; Mar 27(2): 92–8.
71.Sanderson JE. Diastolic heart failure and the extracellular matrix. Int J Cardiol. 1997; Dec 1 62 Suppl 1: S19–21.
72.van den Borne SW, Diez J, Blankesteijn WM, Verjans J, Hofstra L, Narula J. Myocardial remodeling after infarction: the role of myofibroblasts. Nat Rev Cardiol. 2010; Jan 7(1): 30–7.
73.van Amerongen MJ, Bou-Gharios G, Popa E, van Ark J, Petersen AH, van Dam GM, . Bone marrow-derived myofibroblasts contribute functionally to scar formation after myocardial infarction. J Pathol. 2008; Feb 214(3): 377–86.
74.Haudek SB, Xia Y, Huebener P, Lee JM, Carlson S, Crawford JR, . Bone marrow-derived fibroblast precursors mediate ischemic cardiomyopathy in mice. Proc Natl Acad Sci USA. 2006; Nov 28 103(48): 18284–9.
75.Orlic D, Kajstura J, Chimenti S, Bodine DM, Leri A, Anversa P. Bone marrow stem cells regenerate infarcted myocardium. Pediatr Transplant. 2003; 7 Suppl 3: 86–8.
76.Lopez B, Ravassa S, Gonzalez A, Zubillaga E, Bonavila C, Berges M, . Myocardial Collagen Cross-Linking Is Associated With Heart Failure Hospitalization in Patients With Hypertensive Heart Failure. J Am Coll Cardiol. 2016; Jan 26 67(3): 251–60.
77.Bailey AJ. Molecular mechanisms of ageing in connective tissues. Mech Ageing Dev. 2001; May 31 122(7): 735–55.
78.Hartog JW, Voors AA, Bakker SJ, Smit AJ, van Veldhuisen DJ. Advanced glycation end-products (AGEs) and heart failure: pathophysiology and clinical implications. Eur J Heart Fail. 2007; Dec 9(12): 1146–55.
79.Gunja-Smith Z, Morales AR, Romanelli R, Woessner JF, Jr. Remodeling of human myocardial collagen in idiopathic dilated cardiomyopathy. Role of metalloproteinases and pyridinoline cross-links. Am J Pathol. 1996; May 148(5): 1639–48.
80.van Kimmenade RR, Januzzi JL, Jr., Ellinor PT, Sharma UC, Bakker JA, Low AF, . Utility of amino-terminal pro-brain natriuretic peptide, galectin-3, and apelin for the evaluation of patients with acute heart failure. J Am Coll Cardiol. 2006; Sep 19 48(6): 1217–24.
81.Metra M, Nodari S, Parrinello G, Specchia C, Brentana L, Rocca P, . The role of plasma biomarkets in acute heart failure. Serial changes and independent prognostic value of NT-proBNP and cardiac troponin-T. Eur J Heart Fail. 2007; Aug 9(8): 776–86.
82.Zannad F, Alia F, Dousset B, Perez A, Pitt B. Limitation of excessive extracellular matrix turnover may contribute to survival benefit of spironolactone therapy in patients with congestive heart failure: insights from the randomized aldactone evaluation study (RALES). Rales Investigators. Circulation. 2000; Nov 28 102(22):2700–6.
83.Lopez B, Gonzalez A, Ravassa S, Beaumont J, Moreno MU, San Jose G, . Circulating Biomarkets of Myocardial Fibrosis: The Need for a Reappraisal. J Am Coll Cardiol. 2015; Jun 9 65(22): 2449–56.
84.Tziakas DN, Chalikias GK, Stakos D, Chatzikyriakou SV, Papazoglou D, Mitrousi K, . Independent and additive prognostic ability of serum carboxy-terminal telopeptide of collagen type-l in heart failure patients: a multi-marker approach with high-negative predictive value to rule out long-term adverse events. Eur J Prev Cardiol. 2012; Feb 19(1): 62–71.
85.Kato TS, Chokshi A, Singh P, Khawaja T, Iwata S, Homma S, . Markers of extracellular matrix turnover and the development of right ventricular failure after ventricular assist device implantation in patients with advanced heart failure. J Heart Lung Transplant. 2012; Jan 31(1): 37–45.
86.Kupari M, Laine M, Turto H, Lommi J, Werkkala K. Circulating collagen metabolites, myocardial fibrosis and heart failure in aortic valve stenosis. J Heart Valve Dis. 2013; Mar 22(2): 166–76.
87.van Kimmenade RR, Januzzi JL, Jr.. Emerging biomarkers in heart failure. Clin Chem. 2012; Jan 58(1): 127–38.
88.Jones K, Saxon L, Cunningham W, Adams P, Guideline Development G. Secondary prevention for patients after a myocardial infarction: summary of updated NICE guidance. BMJ. 2013; Nov 13 347: f6544.
89.McMurray JJ, Packer M, Desai AS, Gong J, Lefkowitz MP, Rizkala AR, . Angiotensin-neprilysin inhibition versus enalapril in heart failure. N Engl J Med. 2014; Sep 11 371(11): 993–1004.
90.Dickstein K, Cohen-Solal A, Filippatos G, McMurray JJ, Ponikowski P, Poole-Wilson PA, . ESC guidelines for the diagnosis and treatment of acute and chronic heart failure 2008: the Task Force for the diagnosis and treatment of acute and chronic heart failure 2008 of the European Society of Cardiology. Developed in collaboration with the Heart Failure Association of the ESC (HFA) and endorsed by the European Society of Intensive Care Medicine (ESICM). Eur J Heart Fail. 2008; Oct 10(10): 933–89.
91.Tyagi SC, Campbell SE, Reddy HK, Tjahja E, Voelker DJ. Matrix metalloproteinase activity expression in infarcted, noninfarcted and dilated cardiomyopathic human hearts. Mol Cell Biochem. 1996; Feb 9 155(1): 13–21.
92.Wollert KC, Drexler H. The renin-angiotensin system and experimental heart failure. Cardiovasc Res. 1999; Sep 43(4): 838–49.
93.Yarbrough WM, Mukherjee R, Escobar CP, Mingoia JT, Sample JA, Hendrick JW, . Selective targeting and timing of matrix metalloproteinase inhibition in post-myocardial infarction remodeling. Circulation. 2003; Oct 7 108(14): 1753–9.
94.Hudson MP, Armstrong PW, Ruzyllo W, Brum J, Cusmano L, Krzeski P, . Effects of selective matrix metalloproteinase inhibitor (PC-116800) to prevent ventricular remodeling after myocardial infarction: results of the PREMIER (Prevention of Myocardial Infarction Early Remodeling) trial. J Am Coll Cardiol. 2006; Jul 4 48(1): 15–20.
95.Jugdutt Bl, Jelani A, Palaniyappan A, Idikio H, Uweira RE, Menon V, . Aging-related early changes in markers of ventricular and matrix remodeling after reperfused ST-segment elevation myocardial infarction in the canine model: effect of early therapy with an angiotensin II type 1 receptor blocker Circulation. 2010: Jul 27 122(4): 341–51.
96.McAloon CJ, Ali D, Hamborg T, Banerjee P, O’Hare P, Randeva H, . Extracellular cardiac matrix biomarkers in patients with reduced ejection fraction heart failure as predictors of response to cardiac resynchronisation therapy: a systematic review. Open Heart. 2017; 4(2): e000639.
97.Zamilpa R, Lindsey ML. Extracellular matrix turnover and signaling during cardiac remodeling following Ml: causes and consequences. J Mol Cell Cardiol. 2010: Mar 48(3): 558–63.
98.Matsui Y, Jia N, Okamoto H, Kon S, Onozuka H, Akino M, . Role of osteopontin in cardiac fibrosis and remodeling in angiotensin ll-induced cardiac hypertrophy. Hypertension. 2004; Jun 43(6): 1195–201.
99.Mavroidis M, Capetanaki Y. Extensive induction of important mediators of fibrosis and dystrophic calcification in desmin-deficient cardiomyopathy. Am J Pathol. 2002; Mar 160(3): 943–52.
100.Vogel W, Cish CD, Alves F, Pawson T. The discoidin domain receptor tyrosine kinases are activated by collagen. Mol Cell. 1997; Dec 1(1): 13–23.
101.Visse R, Nagase H. Matrix metalloproteinases and tissue inhibitors of metalloproteinases: structure, function, and biochemistry. Circ Res. 2003 May 2:92(8): 827–39.
102.Spinale FC. Myocardial matrix remodeling and the matrix metalloproteinases: influence on cardiac form and function. Physiol Rev. 2007; Oct 87(4): 1285–342.
103.Tyagi SC. Dynamic role of extracellular matrix metalloproteinases in heart failure. Cardiovasc Pathol. 1998: May-Jun 7(3): 153–9.
104.Vilahur C, Juan-Babot O, Pena E, Onate B, Casani L, Badimon L. Molecular and cellular mechanisms involved in cardiac remodeling after acute myocardial infarction. J Mol Cell Cardiol. 2011: Mar 50(3): 522–33.
105.Berk BC, Fujiwara K, Lehoux S. ECM remodeling in hypertensive heart disease. J Clin Invest. 2007: Mar 117(3): 568–75.
Amerikos Argyriou, 1 University of Manchester, Manchester, UK
Mihnea-Alexandru Găman, Editor
About the Author: Amerikos Argyriou is a 3rd year medical student at the University of Manchester, Manchester, UK.
Correspondence: Amerikos Argyriou, Address: University of Manchester, Manchester, UK. Email: americos-g9@hotmail.com
Cite as: Argyriou A. How does the Extracellular Matrix Change in the Setting of Heart Failure?. Int J Med Students. 2018 Sep-Dec;6(3):102-9.
Copyright © 2018 Amerikos Argyriou
This work is licensed under a Creative Commons Attribution 4.0 International License.
International Journal of Medical Students, VOLUME 6, NUMBER 3, December 2018