The Relation between Time since Diagnosis and Awareness about Thalassemia.
Mohammed T. Mutar1, Mustafa Majid1, Ammar Jaleel1, Ali Saad1, Ali Abdulmortafea1, Hashim Talib1
doi: http://dx.doi.org/10.5195/ijms.2019.315
Volume 7, Number 1: 6-10
Received 21 09 2018: Accepted 28 04 2019
ABSTRACT
BackgroundThalassemia is an autosomal recessive disease common in Iraq with a prevalence of 35.7 per 100,000. Beta thalassemia major is a life-threatening condition with many complications which if not managed could cause death at an early age. This cross-sectional study aimed to assess the awareness of parents/caregivers of children with beta-thalassemia major and intermedia, as enhancing awareness is the first and the most important step in all prevention programs.
MethodsWe conducted this study in three thalassemia centers (two in Baghdad and one in Al-Nasiriyah) from July 20th, 2017 to September 20th, 2017. This study involved 193 parents of thalassemic children under the age of 15 who come to the centers frequently for blood transfusion. The study assessed the awareness questionnaire through interviews. Data analysis was performed using the Statistical Package for the Social Sciences program (SPSS) version 24.
ResultsWe found that awareness was low in many aspects. The highest knowledge was for foods that thalassemic patients shouldn’t eat and for the early manifestation signs in thalassemic patients which is 94.8% and 86%, respectively. The lowest knowledge was for the consideration of human immune deficiency virus as a transfusion-transmitted disease and for the chance of having an affected child when both parents are carriers which were 37.3% and 11.9%, respectively.
ConclusionParents have low awareness of thalassemia in many aspects. Thalassemia care centers may need to provide more education for the parents of patients with thalassemia.
Keywords: Thalassemia; Awareness; Prevention; Transmission; Blood Transfusion (Source: MeSH-NLM)..
Thalassemia can be defined as an inherited impairment of hemoglobin synthesis, in which there is a partial or complete failure to synthesize a specific type of globin chains.1 Beta thalassemia major also termed Mediterranean or Cooley’s anemia is the most prevalent form of congenital hemolytic anemia. Beta-thalassemia major is a homozygous state, with either complete absence of beta-chain synthesis (β° thalassemia major) or only small quantities of beta-chains are formed (β + thalassemia major).2 These result in excessive formation of alternate hemoglobin, hemoglobin HbF (alpha 2, gamma 2) and HbA2 (alpha 2, delta 2).3 Hétérozygotes of the disease have thalassemia minor, a condition in which there is usually mild anemia and little or no clinical disability, which may be detected only when iron therapy for mild microcytic anemia does not succeed. Anemia starts appearing within the first 4 to 6 months of life when the switchover from g-chain to β-chain production occurs.1 Thalassemia intermedia is called non-transfusion dependent thalassemia. Patients require less regular transfusions than in thalassemia major and it may take the genotype of thalassemia minor or major. Regular blood transfusions are the main determinant of survival of thalassemia patients, and without this procedure, children with β-thalassemia major will die before the age of three years.4
Beta thalassemia is most common among Mediterranean, Southeast Asians and African descents.5 It is the most common type of hereditary anemia registered in Iraq in 16 thalassemic centers in 2015.4 Both males and females are equally affected and this disorder occurs in approximately 4.4% of every 10,000 live births.6 Also, it was estimated that the prevalence of beta-thalassemia carrier status is 1.5%in the general population.7
Patients with thalassemia major and intermedia are susceptible to many complications, such as marked hepatosplenomegaly which occurs due to excessive red cell destruction (25.8% of thalassemia patients in Iraq have splenomegaly and 4.4% have hepatomegaly), extramedullary hematopoiesis, iron overload, osteoporosis (67.5% in Iraq), osteopenia (9.4% in Iraq) and expansion of bones occurring due to marked erythroid hyperplasia and leading to thalassemic facies and malocclusion of the jaw as a result of changes that occur in the facial bones and soft tissues as well.3-4,8-10 Hemochromatosis in thalassemic patients (major and intermedia) is caused by an iron overload due to repeated blood transfusions and this causes damage to the endocrine organs (aberration in the concentration of various hormones), resulting in a slow rate of growth and development, delayed puberty, diabetes mellitus and damage to the liver and heart (5.3% of them have heart disease) such as heart failure and diastolic dysfunction of the left ventricle and right ventricular dilatation and pulmonary hypertension due to infection and inflammatory processes as well as iron overload.3-4,11-13
Thalassemia is a chronic disease which requires providing care to the patients for their entire life and psychological therapy and education when needed.14 Thus, it is necessary to assess caregivers’ awareness to provide better care for children with thalassemia. Since there is no data available in Iraq about this subject, we conducted this study, aiming to assess their awareness and then compare their knowledge with data from other countries as awareness is a critical step in all prevention programs since prevention programs are inefficient in Iraq compared with other countries where thalassemia is endemic.
This is a cross-sectional study, conducted in three thalassemia centers in two provinces: two in Baghdad (Ibn Albalady and Al-Karama) and one in Al-Nasiriyah. The study was held for two months duration from July 20th, 2017 to September 20th, 2017. The target population was parents or caregivers of affected individuals with thalassemia major or intermedia. A convenient sample of 193 caregivers was selected (162 from Baghdad and 31 from Al-Nasiriyah). Inclusion criteria were: parents/caregivers of patients diagnosed with thalassemia in those centers (depending on hemoglobin-electrophoresis test) and classified into major or intermedia based on their needs of regular transfusion (patients who came regularly to these centers for blood transfusion). Their age limit was 15 years old.
Data was collected from caregivers during an interview using a questionnaire which is self-structured. The questionnaire was designed after reviewing literature.15-17 It was designed under the supervision of a pathologist. After designing the questionnaire, a pilot study was conducted among 25 participants and then a modification was done to develop the final questionnaire. The questionnaire was verified by a statistician for content validity. The questionnaire includes information regarding the socio-demographic status of the patients (age, sex, living address, parental level of education, monthly income), history of consanguineous marriages in the family and previous cases of thalassemia in the family. Their awareness level was assessed depending on their answer about questions regarding: mode of transmission of the disease, carrier status definition, complications of the disease (e.g. organ enlargement and hemochromatosis effects), prevention methods, early diagnosis (first signs and symptoms), their knowledge about bone marrow transplantation as permanent treatment, follow-up (avoiding high iron content food and drugs), as well as the routine check-up of their hemoglobin level, iron level and liver function tests. The awareness score was based on the 22 questions of the questionnaire. Each question was given a score of 4.5 out of a total of 100.
Verbal consent was taken from the caregivers and their profile was confidential. The study was approved by the hospital ethics council and a copy of the approval was provided to the editorial staff of the journal. The data was analyzed using Statistical Package for the Social Sciences program (SPSS) version 24. Descriptive statistics in the form of mean, the standard deviation was used for quantitative data while qualitative data was represented by proportions. One-way analysis of variance (ANOVA), Pearson correlation coefficient was used to test for significant correlation with 95% confidence level, which means a p-value less than 0.05 is statistically significant. Post hoc comparisons using the Bonferroni test were conducted to evaluate pairwise differences among the means.
The study group included 103 male and 90 female patients. The mean age was 92.3 ± 47.8 months (highest age is 180 months and the lowest is 7 months). 85.8% of the patients had thalassemia major and the others had thalassemia intermedia. We noticed that 12% of patients had another blood-related genetic disease: e.g. glucose-6-phosphate dehydrogenase (C6PD) deficiency or sickle cell anemia. The majority of caregivers who brought the patients to the hospital was female (69.9%) and the caregivers’ mean age was 38.7 ± 9.4 years (highest age is 82 and the lowest is 20 years). The parents’ level of education is featured in Table 1. Of the accompanying caregivers, 69.9% were female and 30.1%were male. 65.4%of the caregivers were housewives, 15.2%were unemployed, 9.9% were office workers, 4.7% were working in the military field, 1.6%were working in the education field, 1.6% were working in the medical field and the rest had other jobs. Most of the parents were related (80.6%), which emphasizes the fact that autosomal recessive diseases are common in consanguineous marriages. 59.9% of the caregivers had one affected child, 29.7% had two affected children and 10.4% had three or more thalassemic children. 36.5% of the caregivers had another case in the family and 7.3% had a family member with another genetic blood disease. 53.2% of the parents did the usual premarital tests. The majority of the patients was from Almadina region in Baghdad (19.8%), followed by regions from the periphery of Baghdad (10.2%) and Baghdad Al-Jadida and Al-Adamia (9.1% each). In Al-Nasiriyah, most patients were from Qathaa Al-Nasiriyah (3.7% relative to the whole sample).
Table 1.Education Degree of Parents and Caregivers of Thalassemia Patients.
| Education level | Frequency | Valid percent |
|---|---|---|
| None (Illiterate) | 30 | 15.6 |
| Primary education | 76 | 39.6 |
| Intermediate | 39 | 20.3 |
| Preparatory | 27 | 14.1 |
| Bachelor | 11 | 5.7 |
| Institution | 9 | 4.7 |
| Total | 192 | 100 |
| Missing | 1 | - |
Among the caregivers, 80% have heard about bone marrow transplantation; of those, 39.2% thought that it is a benefic procedure, 29.2% were neutral in their opinion, 25.470 claimed that it is a harmful procedure and could only harm the patient and the remaining 6.2% had no opinion about the procedure. Their knowledge about the diseases that can be transmitted through blood transfusion was as follows: 56.9% agreed that hepatitis can be transmitted via blood transfusion, 37.3% agreed that the Human Immunodeficiency Virus (HIV) can be transmitted via blood transfusion, 25.4% believed that diabetes mellitus can be transmitted via blood transfusion, 16.6% believed that cardiovascular diseases can be transmitted via blood transfusion, 6.4% considered blood transfusion as a safe procedure and the remaining 30.6% did not know if there is any disease that can be transmitted via blood transfusion. When the parents were asked about their action regarding the marriage of their normal son with a carrier or an affected individual, 69.0% refused, 13.1% accepted the marriage to the carrier one only and the remaining 17.9% accepted this idea for both carriers or affected individuals.
Regarding their belief about whether thalassemia major is a dangerous disease or not, 50.3% agreed that thalassemia is a dangerous disease and can affect a child’s life. Meanwhile, 26.7% disagreed with this and said that it can be controlled without any big problems, 14.7% were neutral about this and the remaining 8.3% had no idea. Regarding organ enlargements that may present as complications in thalassemic patients, 55.4% agreed that the liver can be enlarged, 83.470 agreed that the spleen can be enlarged and 3.1% denied any enlargement. Regarding the necessity of iron level lowering drugs, 84.9% ascertained the need for such treatment, 10.9% denied the importance of these drugs and 4.2% did not have any idea about these drugs.
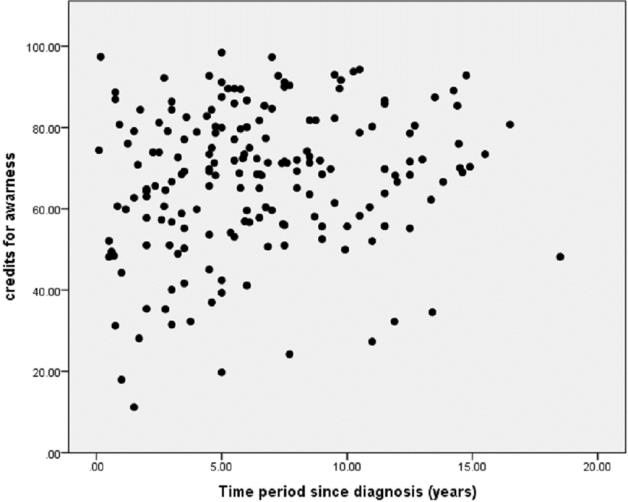
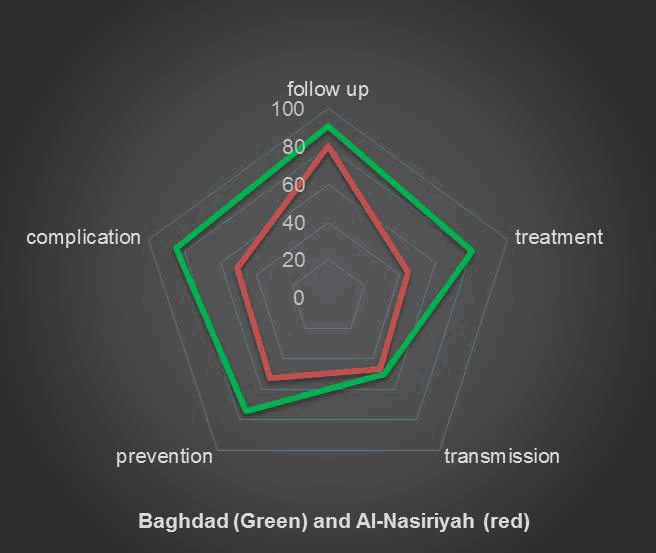
The caregivers’ knowledge about the probability that thalassemia can be transmitted from two carrier parents is illustrated in Table 2. The strength of association between the awareness level and the time of diagnosis of the child, evaluated by the Pearson correlation coefficient (r = 0.18), is shown in Figure 1. The methods of thalassemia prevention, according to our caregivers, are shown in Table 3. We found no relation between the socioeconomic status and the awareness level when compared using one-way analysis of variance (ANOVA) for analysis using a 95% confidence level (p-value=o.1). An association was found between the level of education and level of awareness (p-value<0.001) using one-way analysis of variance (ANOVA) for analysis with 95% confidence level. The awareness level was higher in Baghdad than in Al-Nasiriyah for complications, prevention, follow-up and treatment, while it was the same for the mode of transmission, as shown in Figure 2.
Figure 1.The Relation between Time since Diagnosis and Awareness about Thalassemia.
Comparative Thalassemia Awareness between Baghdad and Al-Nasiriyah.
Caregivers’ Knowledge about the Exact Percent of Thalassemia Inheritance.
| Percentage (answer) | Frequency | Valid percent |
|---|---|---|
| 25% | 22 | 12 |
| 50% | 20 | 10.9 |
| 75% | 31 | 16.8 |
| 100% | 24 | 13 |
| Don't know | 87 | 47.3 |
| Total | 184 | 100 |
| Missing | 9 | - |
Caregivers’ Opinions regarding Thalassemia Prevention Methods.
| Way of prevention | Frequency | Agreement percentage |
|---|---|---|
| Premarital investigations | 159 | 83.2% |
| Avoid consanguineous marriage | 92 | 48.1% |
| Avoid certain food | 28 | 14.7% |
| There is no way for prevention | 21 | 11% |
Overall awareness, ranging from the items of the questionnaire where awareness was the highest to items of the questionnaire where awareness was the lowest, is shown in Table 4.
Table 4.Overall Awareness of Questionnaire Items.
| Question Raised | I agree, Yes | |
|---|---|---|
| 1. | There are many forbidden foods that a thalassemic patient should not eat (like vegetables or meat) | 94.8% |
| 2. | The first sign that thalassemic patients may have is the yellowish discoloration of the skin | 86.0% |
| 3. | Genetic tests are considered as a prevention method of thalassemia | 83.7% |
| 4. | Thalassemic patients may have splenomegaly as a complication of their disease | 83.4% |
| 5. | Did you hear about bone marrow transplantation as a procedure for such patients? | 80.1% |
| 6. | Thalassemia cannot be transmitted by food or being in contact with a patient with thalassemia, do you agree? | 78.2% |
| 7. | Are there any drugs that the thalassemic patients should not take? | 77.7% |
| 8. | Diabetes mellitus cannot be transmitted by blood transfusion | 76.6% |
| 9. | Thalassemic patients should do these tests regularly: hemoglobin, iron blood level and liver and kidney function tests | 72.0% |
| 10. | You are considered as a carrier for thalassemia | 60.0% |
| 11. | Hepatitis can be transmitted via blood transfusion | 56.9% |
| 12. | Thalassemic patients may have hepatomegaly as a complication of their disease | 55.4% |
| 13. | Is thalassemia a genetic disease? | 53.9% |
| 14. | Consanguineous marriage increases the risk of having an affected child | 50.7% |
| 15. | HIV can be transmitted by blood transfusion | 37.3% |
| 16. | The ratio of having an affected child in the case of both carrier parents is 25% in each pregnancy | 11.9% |
As shown in the results section, most patients were brought to thalassemia centers by females, so educational programs should be focused mainly on females. Our results show a very weak linear correlation between the awareness levels and the time period since thalassemia diagnosis which may provide an idea about the continuity of the educational programs provided. Further studies are needed in Iraq to address this subject. Parents did not know that thalassemia is a genetic disease (54%). However, this is considered as a high level of awareness when compared to the awareness in Pakistan according to Kamran Ishfaq et al. where the vast majority had no idea that thalassemia is a genetic disease.18 Some caregivers claim that many factors can be attributed to the etiology of thalassemia (e.g. carcinogens or eating a certain diet) while, in fact, both parents play a role in the transmission of the disease to their children.
Bone marrow transplantation cannot always be affordable for most cases due to financial constraints. Many families send their children overseas to be treated, in particular to Turkey and India, where the procedure costs ($50,000 – $60,000), an amount that many cannot afford. 79.3% of parents know that bone marrow transplantation is a permanent treatment for thalassemia. This percent is high when compared with the study of Chafoor et al. in Pakistan where 75% of the sample denied the presence of permanent treatment.19 25.5% of the sample thought that it is an ineffective treatment while success rates for the cure have been reported as high as 89%-97% worldwide. Baghdad medical city complex and Hiwa Hospital in Sulaymaniyah treat the urgent cases of thalassemia with bone marrow transplants.20-21 Awareness regarding thalassemia complications is relatively good. Their awareness about iron overload was the highest among other complications, suggesting that this is a result of the frequent usage of chelating agents: 68.3% of thalassemic patients in Iraq are on various chelating agents and this also can be attributed to the fact that they have a routine check-up for their iron levels.4
The idea of marriage between a healthy son with a carrier partner was refused by 69.1% of parents because they were still afraid of the disease, even though most of them knew that both parents are required to be carriers for the disease to be transmitted and this showed the need to educate parents about this aspect. Genetic diseases, and especially autosomal recessive diseases, occur more frequently in consanguineous marriages.27 Thus, avoiding such marriages is a known method for preventing many genetic diseases including thalassemia. In our study, we found that 50.8% of parents claim that there is a relationship between consanguineous marriages and the incidence of thalassemia which indicates better knowledge and attitude when compared with the findings of Rudra S et al. study in Bangladesh where only 12% of parents knew that consanguineous marriage is a risk factor for thalassemia.23 Results showed that 83.24% of caregivers agreed that premarital investigation is an effective way to prevent the disease and those became aware of this method after the disease occurred in their family, emphasizing the need for a prevention program. Thalassemia is a preventable disease and some studies have shown that a prevention program consisting of premarital investigations and prenatal screening is a more cost-effective method than treating the patient.24 Today, there is a prevention program implemented in several countries worldwide such as Cyprus, Lebanon, Saudi Arabia, Iran, Qatar, and the United Arab Emirates.23, 25-28 54% of parents considered avoiding consanguineous marriage as a preventive measure which should be done by education among young people who have marriage desires.
In conclusion, awareness in Iraq was generally considered high when compared to other studies conducted in Pakistan and Bangladesh. Parents have low awareness in many aspects, particularly regarding the transmission mode of thalassemia. Finally, we suggest the implementation of educational programs by thalassemia centers to enhance awareness among parents of affected children regarding these issues.
The authors acknowledge Dr. Khitam Razak Al-Khafaji (supervisor of the research) and Ahmed Sameer Alnuaimi and Essam Munir (scientific advisors).
The Authors have no funding, financial relationships or conflicts of interest to disclose.
Conceptualization: MM, MM, AJ, AA, AS, and HT. Methodology: MM, MM, AJ, AA, AS, and HT. Software: MM, MM, AJ, and AS. Validation: MM, MM, AJ, and AS. Formal Analysis: MM, MM, AJ, and AS. Data Curation: MM, MM, AJ, AA, AS., and HT. Investigation: MM, MM, AJ, AA, AS, and HT. Resources: MM, MM, AJ, AA, AS, and HT. Writing – Original Draft: MM, MM, AJ, AA, and HT. Writing – Review & Editing: MM, MM, AJ, and AS. Visualization: MM, MM, AJ, AA, AS, and HT. Supervision: MM, MM, and AJ. Project Administration: MM, MM, AJ, AA, AS, and HT.
1.Mohan H. Textbook of Pathology. 7 ed. New Delhi: Jaypee Brothers Medical Publishers; 2015.
2.Colledge N, Davidson S, Ralston S, Walker B. Davidson's principles and practice of medicine. 21 ed. Edinburgh, New York: Churchill Livingstone/Elsevier, 2010. P. 1029–1030.
3.Modell B, Darlison M. Global epidemiology of haemoglobin disorders and derived service indicators. Bull World Health Organ. 2008;86(6):48o–487.
4.Kadhim KA, Baldawi KH. Lami FH. Prevalence, Incidence, Trend, and Complications of Thalassemia in Iraq. Hemoglobin. 2017 May;41(3):164–168.
5.Muncie HL Jr, Campbell J. α And β thalassemia. Am Fam Physician. 2009;80(4):339–344.
6.Saxena R, Banerjee T, Aniyery RB. Thalassemia and its Management during Pregnancy. World I Anemia. 2017;1(1):5–17.
7.Vichinsky EP. Changing patterns of thalassemia worldwide. Ann NY Acad Sci. 2005;1054:18–24.
8.AL Jadit SM, Jalal MZ, AL Ghreer MF, AL Hamdani MS, AL Omaree WR. Osteoporosis in Iraqi patients with thalassemia. Arthritis Res Ther. 2002;14(Suppl 1):P4.
9.Karakas S, Tellioglu AM, Bilgin M, Omurlu IK, Caliskan S, Coskun S. Craniofacial Characteristics of Thalassemia Major Patients. Eurasian J Med. 2016 Oct;48(3):204–208.
10.Amini F, Jafari A, Eslamian L, Sharifzadeh S: A cephalometric study on craniofacial morphology of Iranian children with beta-thalassemia major. Orthod Craniofacial Res, 2007;10:36–44
11.Abdulzahra MS, Al-Hakeim HK, Ridha MM. Study of the effect of iron overload on the function of endocrine glands in male thalassemia patients. Asian J Transfus Sci. 2011 Jul-Dec;5(2):127–131.
12.Kremastinos DT, Farmakis D, Aessopos A, Hahalis G, Hamodraka E, Tsiapras D, . β-Thalassemia Cardiomyopathy. Circulation: Heart Failure. 2010;3:451–458.
13.Arif F, Fayyaz J, Hamid A. Awareness among parents of children with thalassemia major. J Pak Med Assoc. 2008;58:621–3.
14.Anie KA, Massaglia P. Psychological therapies for thalassaemia. Cochrane Database Syst Rev. 2014 Mar 6;(3):CD002890.
15.Jenn NC. Designing A Questionnaire. Malays Fam Physician. 2006;1(1):32–35.
16.Boynton P. M, Greenhalgh, T. Selecting, designing, and developing your questionnaire. BMJ. 2004; 328:1312.
17.Leung WC. How to conduct a survey. Student BMJ. 2001 May;9:143–5.
18.Ishfaq K, Ahmad T, Naeem SB, Ali J, Zainab F. The knowledge of parents having thalassemia child. Isra Medical Journal, 2016;8(2):79–81.
19.Bilal M, Saleem M, Mustafa G, Naveed S. Level of Awareness about Thalassemia among Parents of Thalassaemic Children. Journal of Rawalpindi Medical College (JRMC); 2016;20(3):209-21 1.
20.Lucarelli G, Galimberti M, Polchi P, Angelucci E, Baronciani D, Giardini C, . Marrow transplantation in patients with thalassemia responsive to iron chelation therapy. N Engl J Med. 1993;329:840–44.
21.Hammadi AM, Azeez WA, Jasim FH, Alshammary N, Sewan AD, Alrawaq K, . First Report on Stem Cell Transplant from Iraq. Exp Clin Transplant. 2017 Feb;15(Suppl 1):133–135.
22.Machado T, Bomfim T, Souza L, Soares N, Santos F, Acosta A, . Types of marriages, population structure and genetic disease. Journal of Biosocial Science. 2013;45(4):46l–470.
23.Rudra S, Chakrabarty P, Hossain MA, Ripon MJ, Rudra M, Mirza T. Awareness among Parents of β-Thalassemia Major Patients Regarding Prenatal Diagnosis and Premarital Screening in Day Care Centre of Transfusion Medicine Department. Mymensingh Med J. 2016 Jan;25(1):1 2-7.
24.Ahmadnezhad E, Sepehrvand N, Jahani F. Evaluation and Cost Analysis of National Health Policy of Thalassaemia Screening in West-Azerbaijan Province of Iran. Int J Prev Med. 2012 Oct;3(1o):687–692.
25.Kalokairinou EM. The experience of β-thalassaemia and its prevention in Cyprus. Med Law 2017:26: 291–307.
26.Inati A, Zeineh N, Isma'eel H, Koussa S, Gharzuddine W, Taher A. β-thalassemia: The Lebanese experience. Clin Lab Haematol 2006;28:217–227.
27.Samavat A, Modell B. Iranian national thalassaemia screening programme. BMJ. 2004;329:1134–1137.
28.Antonio C, Kan YW. The Prevention of Thalassemia. Cold Spring Harb Perspect Med. 2013 Feb 1;3(2):a011775.
Mohammed T. Mutar, 1 Medical Student, Baghdad University College of Medicine
Mustafa Majid, 1 Medical Student, Baghdad University College of Medicine
Ammar Jaleel, 1 Medical Student, Baghdad University College of Medicine
Ali Saad, 1 Medical Student, Baghdad University College of Medicine
Ali Abdulmortafea, 1 Medical Student, Baghdad University College of Medicine
Hashim Talib, 1 Medical Student, Baghdad University College of Medicine
Mihnea-Alexandru Găman, Editor
About the Author: Muhammed Tareq Mutar is a fourth-year medical student at Baghdad University College of Medicine, Iraq of a six-year program. He is a member of the British Nuclear Medicine Society and a reviewer in Cogent Medicine journal.
Correspondence: Mohammed Tareq Mutar, Address: Baghdad University College of Medicine, Iraq. Email: muhammed.tariq64@gmail.com
Cite as: Mutar MT, Majid M, Jaleel A, Saad A, Abdulmortafea A, Talib H. Awareness among Parents of Beta Thalassemia Major and Intermedia Patients in Three Centers in Baghdad and Al-Nasiriyah, Iraq in 2017. Int J Med Students. 2019;7(1):6-10.
Copyright © 2019 Mohammed T. Mutar, Mustafa Majid, Ammar Jaleel, Ali Saad, Ali Abdulmortafea, Hashim Talib
International Journal of Medical Students, VOLUME 7, NUMBER 1, April 2019