Case Report
Polymyositis as a Rare Musculoskeletal Manifestation of Chronic Graft-Versus-Host
Disease: A Case Report of a 33-Year-Old Patient
David Ben-Nun1
doi: http://dx.doi.org/10.5195/ijms.2020.487
Volume 8, Number 1: 36-40
Received 28 03 2020:
Accepted 10 04 2020
ABSTRACT
Background:
Musculoskeletal manifestations of chronic Graft-Versus-Host Disease (GVHD) are rare
and unfamiliar to most clinicians. Here we discuss the pathophysiology of chronic
GVHD, current treatments and direction of research for improved therapy and prophylaxis
and focus on the common and less common musculoskeletal complications of this unfortunately
prevalent and burdensome disease.
The Case:
This is a case report of a 33-year-old male with a past medical history of Acute Myeloid
Leukemia (AML) who presented with a 1-month history of generalized, proximal weakness
and was found to have polymyositis secondary to chronic GVHD. Elicitation of further
history showed that the patient had had multiple manifestations of both acute and
chronic graft-versus-host disease in the two years following hematopoietic stem cell
transplantation (HSCT).
Conclusion:
It is important for clinicians to be familiar with polymyositis secondary to chronic
GHVD, which may appear in patients more than 100 days following allogenic HSCT and
typically presents as diffuse, generalized myopathy with preserved sensation and elevated
CPK and aldolase. The current mainstay of treatment is glucocorticoids with or without
a calcineurin inhibitor, however due to the side effects associated with long term
treatment, more effective prophylactic and therapeutic treatments are needed to address
this and other manifestations of chronic GVHD.
Keywords:
Polymyositis;
Graft-Versus-Host Disease;
Muscular Diseases;
Acute Myeloid Leukemia;
Hematopoietic Stem Cell Transplantation (Source: MeSH-NLM).
Introduction
Graft-Versus-Host Disease (GVHD) is a pathological process that occurs when immune
cells from a donor graft recognize host antigens as foreign and trigger an immune
response. GVHD is the most common life-threatening complication of allogenic hematopoietic
stem cell transplantation (HSCT) and is classified into acute and chronic based on
the length of time after which the immune response occurs following transplantation.1 Acute GVHD is considered any manifestation of graft T cell immune activation within
the first 100 days following HSCT.2 Typical signs and symptoms of acute GVHD include a maculopapular rash, hyperbilirubinemia
with jaundice, nausea and vomiting, anorexia, and watery or bloody diarrhea with crampy
abdominal pain.3 By some estimates, incidence of acute GVHD may be as low as 9% or up to 50% of patients
who undergo HSCT from Human Leukocyte Antigen (HLA)-matched sibling donors, which
indicates that it is a significant medical problem especially given the fact that
HSCT is a relatively common procedure.4–6 Between the years 2013-2017, the Center for International Blood and Marrow Transplant
Research reported that 106,112 HSCT procedures were carried out in the United States.
Out of that total, approximately 43,962 were allogeneic transplants, which is an average
of 8,792 allogeneic HSCTs annually.7
Chronic GVHD was first described in 1978 and has been defined as any alloimmunity
that results in clinical manifestations that occur more than 100 days post HSCT.(2)
Chronic GVHD occurs in 30 to 70% of patients who have undergone allogeneic HSCT of
nonmanipulated donor grafts and have received standard prophylaxis with a calcineurin
inhibitor and an antimetabolite.(1) Chronic GVHD can have protean manifestations that
range from fibrotic skin disease resembling systemic sclerosis, bronchiolitis, salivary
and lacrimal gland disease, and eosinophilic fasciitis.8
The pathophysiology of chronic GVHD is believed to involve three phases: early inflammation
and tissue injury, chronic inflammation and dysregulated immunity, and aberrant tissue
repair often accompanied by fibrosis.(9) In the first phase, there is translocation
of bacteria and fungi across epithelial tight junctions, which are believed to be
made more porous as a result of tissue damage that may occur following cytotoxic cell
conditioning with chemotherapy or radiotherapy during the process of HSCT. This porosity
leads to the release of immunogenic molecules that are not normally found in the extracellular
space, which triggers clonal expansion of B cells and differentiation of T cells into
type 1, type 2, and type 17 helper T-cells. This expansion of T cells causes a concomitant
increase in the amount of auto-reactive T cells that escape immune regulation both
in the thymus and in the periphery. Autoreactive T cells, along with toxic effects
of the conditioning regimen, prophylaxis with calcineurin inhibitors, and immunoglobulin
deposition lead to further thymic injury which further degrades the immune system's
ability to filter out autoreactive immune cells in the thymus. In the final phase,
activated macrophages secrete fibrotic growth factors such as Transforming Growth
Factor Beta (TGF-β) and Platelet-Derived Growth Factor (PDGF), which stimulate fibroblasts
to lay down extracellular collagen and causes widespread fibrosis and sclerosis in
various organs.1
In 2005, the National Institutes of Health (NIH) compiled a formal set of guidelines
regarding diagnosis of chronic GVHD, which were updated in 2014, that stipulate that
the diagnosis of chronic GVHD requires “at least 1 diagnostic manifestation of chronic
GVHD or at least 1 distinctive manifestation, with the diagnosis confirmed by pertinent
biopsy, laboratory tests, or radiology in the same or another organ” along with distinction
from acute GVHD and exclusion of other possible causes.4 The mainstay of treatment of chronic GVHD requiring systemic treatment is and has
been glucocorticoids, along with a calcineurin inhibitor. If patients require additional
therapy following the prolonged use of glucocorticoids, secondary agents including
rituximab, cyclophosphamide, imatinib, mycophenolate mofetil, IL-2, extracorporeal
phonophoresis, methotrexate, bortezomib and as well as other novel immunosuppressive
treatments are used.1,10
Musculoskeletal manifestations that result from chronic GVHD are uncommon and likely
to be missed by many clinicians who are unaware of this entity. The goal of reporting
on the case below is to emphasize the importance of being aware of the presentation,
diagnostic guidelines and recommended treatment of polymyositis secondary to chronic
GVHD in patients who have undergone HSCT.
The Case
Here we report on the case of a 33-year-old male living in central Israel with a past
medical history of Acute Myeloid Leukemia (AML) diagnosed two years prior who presented
to his local hospital with a 1-month history of generalized weakness that was more
pronounced in the lower body. The patient's chief complaint was that he was having
difficulty accomplishing tasks of daily living such as lifting items around the house,
getting out of bed, preparing food and driving. He had initially gone to see a neurologist
in his community, who noted that he had profound upper and lower body weakness and
referred him to the local hospital. The patient stated that the weakness had begun
to progress more rapidly in the 10 days leading up to his presentation and that he
now needed to use crutches in order to walk. He reported no sensory deficits or urinary
incontinence. He also denied a family history of neurological or musculoskeletal disease,
significant history of substance abuse, recent notable travel or sick contacts or
occupational exposures to toxic chemicals.
The patient had been generally healthy prior to his diagnosis of AML two years prior.
At that time, he had presented with complaints of fever, weakness, vertigo and headache
for one week. Following an extensive work up, the patient was diagnosed with FMS-like
tyrosine kinase 3-internal tandem duplication (FLT3-ITD) positive AML, which is named
for its similarity to a tyrosine kinase that binds macrophage or monocyte colony-stimulating
factor and is encoded by the Feline McDonough Sarcoma (FMS) oncogene.11 He was treated with a “7+3” regimen of daunorubicin and cytarabine plus midostaurine
as well as prophylactic antibiotics and steroids. The patient ultimately underwent
allogenic HSCT two months after his initial presentation with a donor who was a 10/10
match (both alleles matched at HLA loci A, B, C, DRB1, and DQB1). One month following
his HSCT, the patient was hospitalized with a febrile illness and found to have elevated
CMV titers as well as elevated AST and ALT enzymes. He was treated at the time for
a suspected viral infection with valganciclovir, piperacillin/tazobactam and prednisone.
The patient later developed facial skin rashes and oral ulcers and his prednisone
dose was increased. Soon afterward, he also suffered from a febrile illness with respiratory
symptoms including cough and slight oxygen desaturation. He underwent bronchoscopy
and a biopsy was taken from his respiratory tract, which was later found to be positive
for Corynebacterium and he was treated with antibiotics. Later that same year, the
patient presented with a complaint of gross hematuria and he was again prescribed
prednisone and the hematuria eventually resolved.
At the patient's presentation for a chief complaint of generalized weakness, he was
taking voriconazole, acyclovir, and trimethoprimsulfamethoxazole for antibacterial
and antiviral prophylaxis as well as sorafenib for maintenance AML treatment. He was
tapering his dose of prednisone and currently taking 5mg daily, which was a decrease
from 60mg daily a few months prior.
At the hospital, the patient's vital signs showed that he was afebrile with a regular
pulse of 85, a blood pressure of 107/55 and a respiratory rate within normal limits.
The patient's extraocular muscle movements and visual fields were intact. Both arms
and legs were hypotonic and patient could not hold them up against gravity, however
no pronation was noticed. Arm muscle strength was rated as 4+/5 bilaterally both proximal
and distal. Wrist flexion was rated as 5/5. In the lower body, hip flexion was rated
as 3/5 bilaterally, knee flexion was 4+/5 bilaterally, knee extension was 4+/5 bilaterally
and foot plantar flexion and dorsiflexion was rated as 5/5 bilaterally. The patient's
reflexes were 2+ throughout without any pathological reflexes. Superficial touch was
intact in upper and lower body. The patient's gait was narrow-based with left foot
dragging and he required a crutch in order to move around in his hospital room.
Laboratory results showed that the patient had a macrocytic anemia of 12.3 g/dL with
a MCV of 99.4 (normal 80-94), thrombocytopenia of 90 K/micl (normal 130-400), leukocytosis
of 13.32 K/micl (normal 4.8-10.8) with eosinophilia of 4.3 K/micl (normal 0.0-0.8).
All of these abnormal blood test findings were confirmed to have been noted on prior
labs. The patient had CRP a 1.86 mg/L (normal 0.0 – 5.0) and ESR of 29 (normal 0.0
– 8.0). Basic metabolic and coagulation panels were within normal limits. Patient's
CPK and TSH were within normal limits. A lumbar puncture was performed which revealed
an opening pressure of 12 cm of H2O, 1 WBC/uL, no erythrocytes, glucose of 47 mg/dL
and no xanthochromia. A flow cytometry test was negative for any abnormal cell populations
in CSF and CSF culture and a Biofire panel for various microbiological pathogens was
also negative. Patient serum and CSF was checked for antibodies including ANA, anti-dsDNA,
anti-Jo1, anti-Scl-70, anti-voltage-gated calcium channel, anti-acetylcholine receptor,
anti-GM-1 among others and were all negative. Hepatitis and HIV tests were also negative.
The patient was sent for a non-contrast head CT which showed a minor lesion in the
right pons that was interpreted as an old finding that was likely an artifact.
The patient was also sent for electromyography (EMG), which revealed no spontaneous
activity upon needle insertion and low amplitude, polyphasic motor unit action potentials
with decreased duration along with increased recruitment in all nerves including the
median, ulnar, peroneal, tibial and axillary nerves. Nerve conduction studies showed
distal motor latency and conduction velocity were preserved for both sensory and motor
components.
The patient was treated with 60mg of methylprednisolone IV and was referred to hematology
for treatment of presumed polymyositis secondary to chronic GVHD. Muscle biopsy was
also recommended in order to confirm the working diagnosis but deferred since it seemed
reasonable to assume this was a manifestation of chronic GVHD given the patient's
history and findings. Following a stay in the hematooncology department, the patient's
dose of steroids was changed to 50mg daily of prednisone. The patient was instructed
to continue taking prophylactic antibiotics and schedule a follow up appointment with
his hematologist for continued treatment of chronic GVHD manifestations.
Discussion
This report details the case of a patient with a history of FLT3-ITD positive AML
and acute and chronic GVHD who presented with progressive weakness of one month's
duration and was diagnosed with presumed polymyositis secondary to chronic GVHD. According
to the NIH Consensus Criteria on Chronic GVHD, polymyositis is a recognized diagnostic
finding in chronic GVHD (Figure 1) and, in this case, it was confirmed via EMG, which clearly demonstrated a myopathic
pattern in multiple nerves in the upper and lower body. Other causes of myopathy including
prolonged steroid use, auto-immune disease, hypokalemia, hypothyroidism, infection
and otherwise were ruled out and the patient's presentation timeframe of more than
100 days post HSCT excluded acute GVHD as a potential cause.
Figure 1.
Section of Scoring Card from NIH Chronic Graft Versus Host Disease Criteria Relevant
to Musculoskeletal Manifestations.
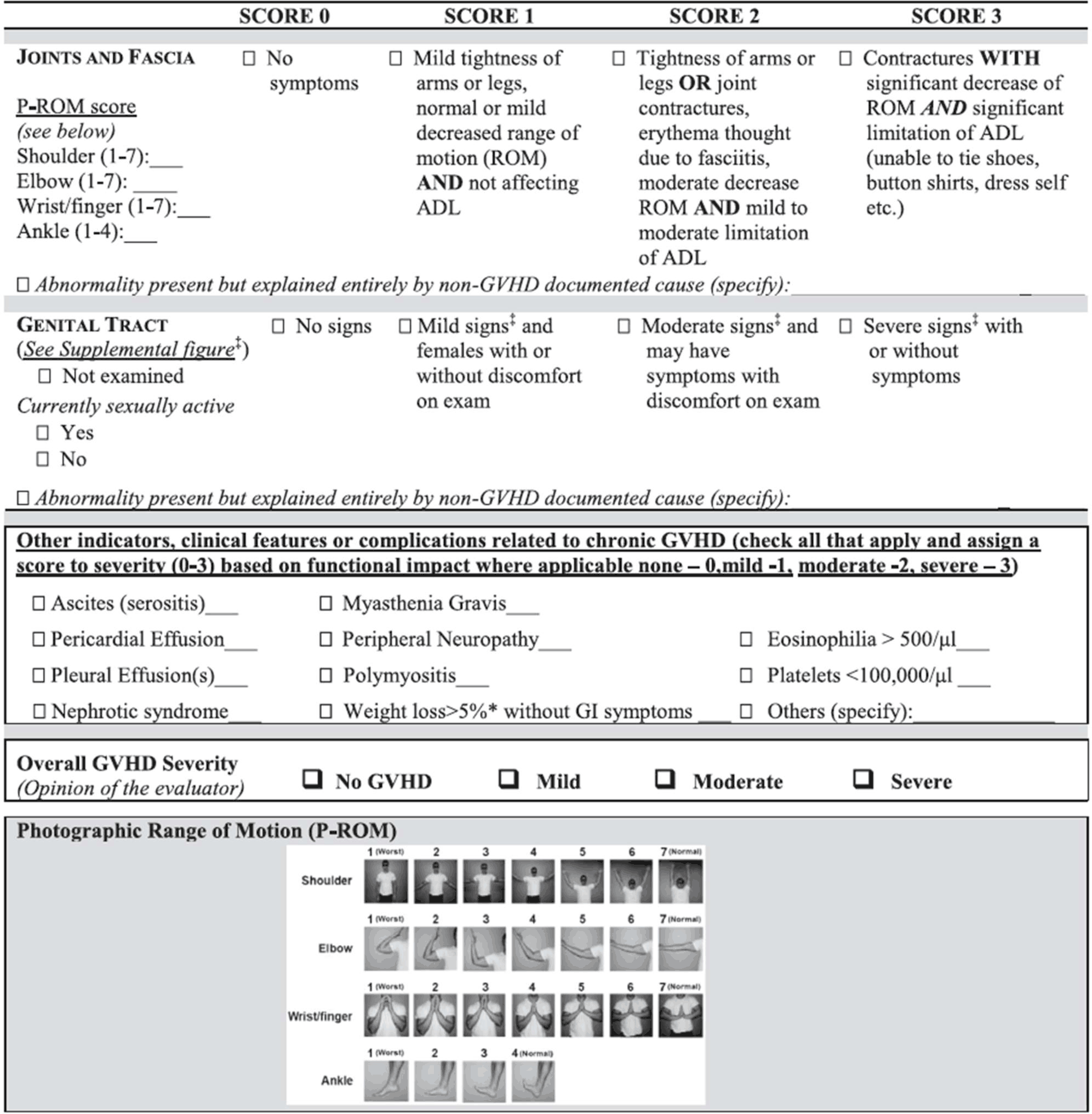
Legend. Section of organ system scoring card relevant to musculoskeletal complications of
chronic GVHD as drafted by The National Institutes of Health Consensus Development
Project on Criteria for Clinical Trials in Chronic Graft-versus-Host Disease in 2014.
The scoring card focuses mostly on musculoskeletal changes related to joint contractures,
fasciitis and their related manifestations with a photographic range of motion scale
provided for ranking perceived joint range of motion. There is also a box that can
be ticked with a related severity scale of 1-3 in case of noted polymyositis.15
Musculoskeletal manifestations of chronic GVHD are very rare and when present do not
usually manifest as myositis. In a retrospective chart review of all cases of myositis
that developed in 7161 patients who underwent HSCT at the Fred Hutchinson Cancer Research
Center in Seattle, Washington between 1969 and 1999, only 12 patients out of 1859
(0.6%) who developed chronic GVHD also developed myositis between 7 and 55 months
after HSCT.12 In another case series of 381 patients with chronic GVHD, Parker at el. reported
that myositis occurred in 11 patients out of 318 (3.5%).13 The incidence of myositis in the first study at the Hutchinson Cancer Center was
calculated to be 23 per 100,000 person-years for all HSCT patients and 49 per 100,000
person-years for HSCT patients who had previously reported chronic GVHD manifestations.12 For a comparison of prevalence in the general population, in a large study that looked
at 35 million commercially insured individuals in the US, the incidence of idiopathic
inflammatory myositis (including dermatomyositis, polymyositis and sporadic inclusion
body myositis), was found to be between 5.8 to 7.9 per 100,000 person-years from 2003-2008.14
In patients who do develop musculoskeletal complications of chronic GVHD, the most
common manifestation is fasciitis that is characterized by sclerosis of the overlying
skin and subcutaneous tissues causing joint contractures, edema and restricted range
of motion.15 Figure 1 shows the section of the scoring card that is pertinent to musculoskeletal manifestations
provided by the National Institutes of Health to rate the severity of chronic GVHD.
In that section, a scoring system is shown that allows clinicians to rate the severity
of joint contractures and to tick a box if polymyositis, peripheral neuropathy and/or
myasthenia gravis are present and, if so, to indicate the degree of severity. Besides
evaluating for joint contractures, clinicians caring for patients with musculoskeletal
symptoms of chronic GVHD should always monitor a patient's respiratory rate and oxygen
saturation since there are reports of respiratory failure as a result of weakness
of the respiratory muscles in chronic GVHD.16
Another etiology of myositis that was considered in this patient was medication-induced
myositis. The patient has been regularly taking sorafenib, a tyrosine kinase inhibitor
that has been shown to increase event-free survival in patients younger than 60 years
old with FLT3-ITD positive AML albeit with increased toxicity.17 While Sorafenib has been reported to cause myositis in at least one case report,
however it was not believed to be the culprit in this patient's case since the patient
had been taking the drug for several months before the onset of weakness and myopathic
symptoms appear to be a relatively rare side effect.18 As a result, the patient was instructed to continue taking sorafenib as maintenance
for AML.
This patient presented with normal levels of CPK, which may make this presentation
unusual since in one review of 36 cases of polymyositis due chronic GVHD, it was reported
that the majority of patients presented with elevated CPK and aldolase levels.19 Aldolase was not measured in this patient, so unfortunately we cannot know if it
was elevated or not. As for the normal level of CPK, while this enzyme typically parallels
disease activity in most patients with myositis, it may be normal or only slightly
elevated in different types of myositis.20 This patient did have a history of acute GVHD, which may or may not be present in
common presentations of this disease. In the same case series that reported data on
elevated CPK, Couriel et al. found that about half of the patients did not have any
history of an episode of acute GVHD prior to their presentation with symptoms of polymyositis.19 This also means that a significant percentage of patients are likely to present with
myositis without any prior indications of graft-versus-host disease.
In muscle biopsies taken in cases of myositis secondary to chronic GVHD, one should
expect the same findings as in idiopathic polymyositis, namely perimysial and endomysial
infiltrate of CD4+ and CD8+ T cells in the myofibrils.19 This is confirmed in a report by Takahashi et al., who reported CD4+ and CD8+ T cells
in the perimysial infiltrate and primarily CD8+ infiltrate in the endomysial layer
in a 22-year-old patient with polymyositis secondary to chronic GVHD.21 Examples of pathology slides showing expected findings can be seen in Figure 2 and Figure 3.
Figure 2.
Lymphocytic Infiltrate in Muscle Tissue.
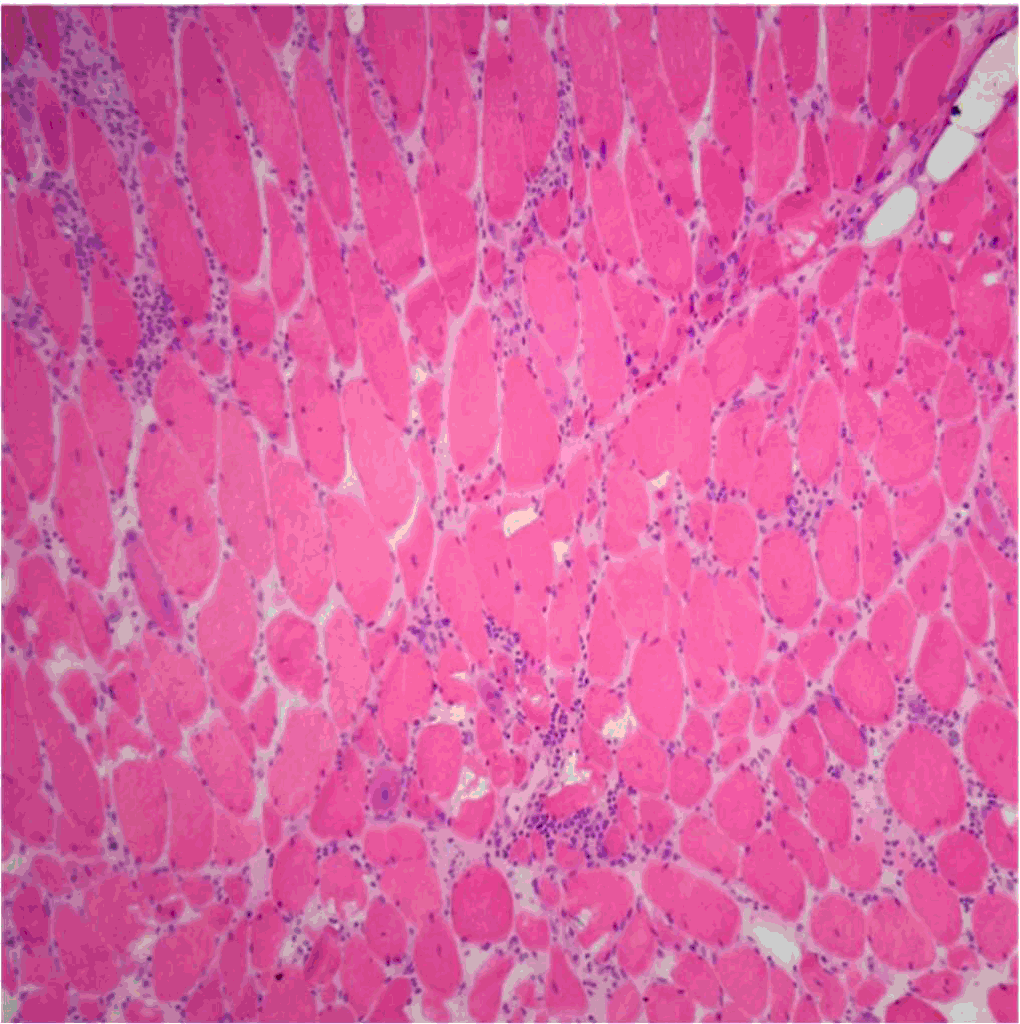
Legend; An H&E section of a muscle tissue showing infiltration of lymphocytes as well as
myopathic features including smaller, rounded myofibers with increased internal nuclei.
Courtesy of Meggen Walsh, D.O., M.S.-P.A.22
Figure 3.
CD8+ T Cell Infiltrate in Muscle Tissue
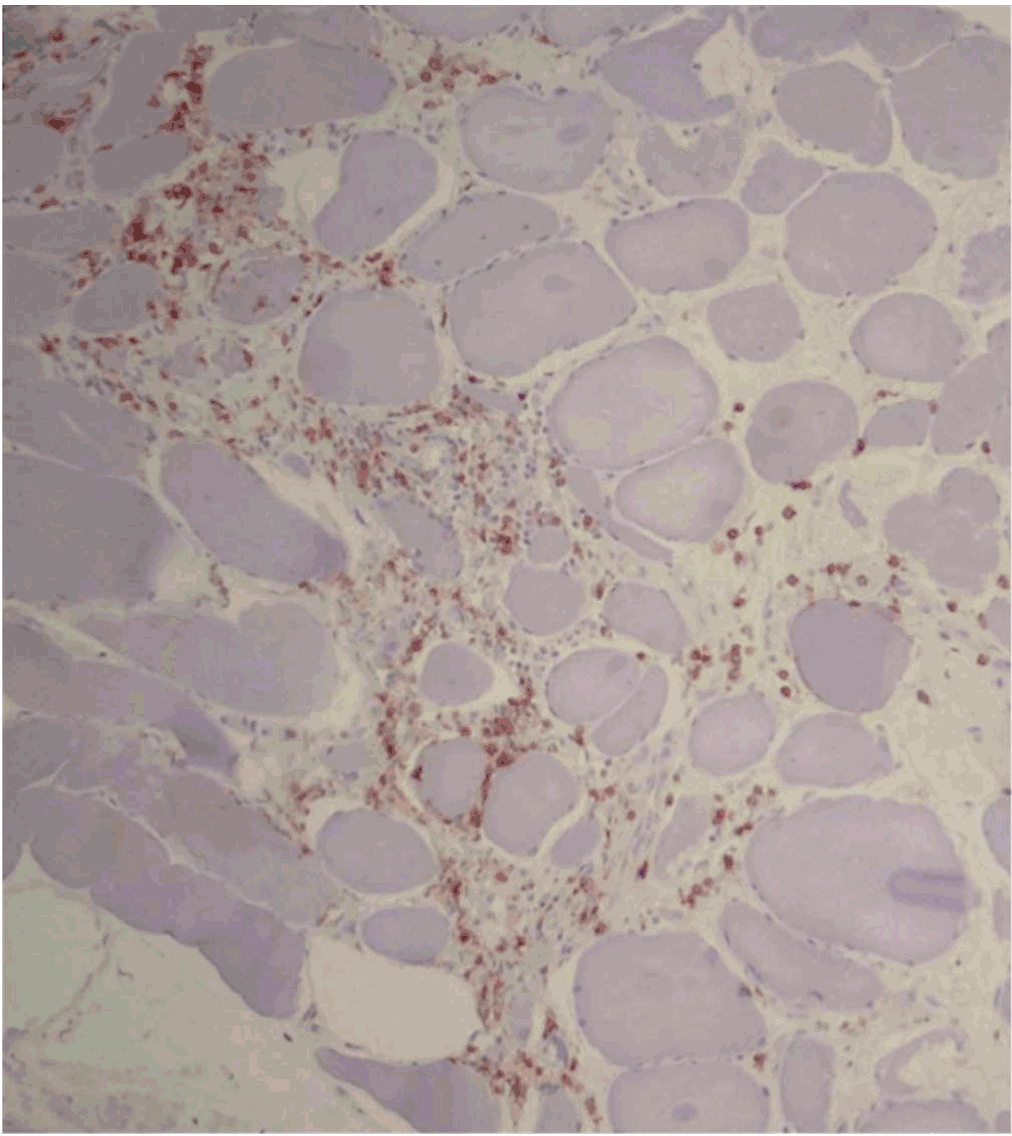
Legend: Immunostaining of muscle from H&E section showing that infiltrate is composed of
CD3+ CD8+ T cells. Courtesy of Meggen Walsh, D.O., M.S.-P.A.22
This case shows that it is important for clinicians to be familiar with the phenomenon
of polymyositis as a manifestation of chronic GVHD, especially since the etiology
of myopathy in these patients may easily be mistaken for something else since they
often suffer from multiple co-morbidities and are often on immunosuppressive therapy
as well as other medications. Furthermore, more research is necessary to find more
effective prophylactic and therapeutic treatments for chronic GVHD that possess a
lighter side effect burden with long-term use. Current directions of investigation,
which include impeding B cell signaling and germinal center formation, increasing
regulatory T cells populations, targeting pro-fibrotic interleukins and T and B cell
recruiting chemokines have shown some promise but require more patients and clinical
trials to be further validated.1
Acknowledgments
I would like to thank the Neurology Department at Beilinson Rabin Medical Center in
Petah Tikva, Israel for providing access to patient information for the purposes of
creating this manuscript.
Conflict of Interest Statement & Funding
The Authors have no funding, financial relationships or conflicts of interest to disclose.
Author Contributions
Conceptualization: DBN. Methodology: DBN. Software: DBN. Validation: DBN. Formal Analysis:
DBN. Data Curation: DBN. Investigation: DBN. Resources: DBN. Writing – Original Draft:
DBN. Writing – Review & Editing: DBN. Visualization: DBN. Supervision: DBN. Project
Administration: DBN.
References
1.Zeiser R, Blazar BR. Pathophysiology of chronic graft-versus-host disease and therapeutic targets. N Engl J Med. 2017 Dec 28; 377(26):2565–79.
2.Lee SJ. Classification systems for chronic graft-versus-host disease. Blood. 2017 Jan5; 129 (1):30–7.
3.Zeiser R, Blazar BR. Acute graft-versus-host disease - Biologic process, prevention, and therapy. N Engl J Med. 2017 Nov30; 377 (22):2167–79.
4.Filipovich AH, Weisdorf D, Pavletic S, Socie G, Wingard JR, Lee SJ. National Institutes of Health Consensus Development Project on criteria for clinical
trials in chronic graft-versus-host disease: I. diagnosis and staging working group
report. Biol Blood Marrow Transplant. 2005 Dec; 11 (12):945–56.
5.Veltri L, Regier M, Cumpston A, Leadmon S, Tse W, Craig M. Incidence and pattern of graft-versus-host disease in patients undergoing allogeneic
transplantation after nonmyeloablative conditioning with total lymphoid irradiation
and antithymocyte globulin. Bone Marrow Res. 2013, 2013:414959.
6.Lee SE, Cho BS, Kim JH, Yoon JH, Shin SH, Yahng SA. Risk and prognostic factors for acute GVHD based on NIH consensus criteria. Bone Marrow Transplant. 2013 Apr; 48 (4):587–92.
7.Center for International Blood and Marrow Transplant Research. Total Number of HCTs Performed in the United States and Reported to Center for International
Blood and Marrow Transplant Research - By Donor Type: 2013-2017. Health Resources and Services Administration. Available from: https://bloodstemcell.hrsa.gov/sites/default/files/bloodstemcell/data/transplant-activity/transplants-donor-type.pdf. CitedMar16,2020.
8.Shlomchik WD. Graft-versus-host disease. Nat Rev Immunol. 2007 May; 7 (5):340–52.
9.Cooke KR, Luznik L, Sarantopoulos S, Hakim FT, Jagasia M, Fowler DH. The Biology of Chronic Graft-versus-Host Disease: A Task Force Report from the National
Institutes of Health Consensus Development Project on Criteria for Clinical Trials
in Chronic Graft-Versus-Host Disease. Biol Blood Marrow Transpl. 2017 Feb; 23 (2):211–34.
10.Vogelsang GB. How I treat chronic graft-versus-host disease. 2001 Mar 1; 97 (5):1196–201.
11.El-Gamal MI, Anbar HS, Yoo KH, Oh C-H. FMS Kinase Inhibitors: Current Status and Future Prospects. Med Res Rev. 2013 May; 33 (3):599–636.
12.Stevens AM, Sullivan KM, Nelson JL. Polymyositis as a manifestation of chronic graft-versus-host disease. Rheumatology. 2003 Jan; 42 (1):34–9.
13.Parker P, Chao NJ, Ben-Ezra J, Slatkin N, Openshaw H, Niland JC. Polymyositis as a a Manifestation of Chronic Graft-Versus-Host Disease. Medecine (Baltimore). 1996 Sep; 75 (5):279–85.
14.Furst DE, Amato AA, Iorga Ş R, Gajria K, Fernandes AW. Epidemiology of adult idiopathic inflammatory myopathies in a U.S. managed care plan. Muscle and Nerve. 2012 Jan; 45 (5):676–83.
15.Jagasia MH, Greinix HT, Arora M, Williams KM, Wolff D, Cowen EW. National Institutes of Health Consensus Development Project on Criteria for Clinical
Trials in Chronic Graft-versus-Host Disease: I. The 2014 Diagnosis and Staging Working
Group Report. Biol Blood Marrow Transplant. 2015 Mar; 21 (3):389–401.
16.Stephenson AL, Mackenzie IRA, Levy RD, Road J. Myositis associated graft-versus-host-disease presenting as respiratory muscle weakness. Thorax. 2001 Jan; 56 (1):82–4.
17.Röllig C, Serve H, Hüttmann A, Noppeney R, Müller-Tidow C, Krug U. Addition of sorafenib versus placebo to standard therapy in patients aged 60 years
or younger with newly diagnosed acute myeloid leukaemia (SORAML): A multicentre, phase
2, randomised controlled trial. Lancet Oncol. 2015 Dec; 16 (16):1691–9.
18.Diaz-Sanchez A, Rodriguez-Salas N, Aramendi T, Balbin E. Myositis due to Sorafenib intake in a patient with hepatocellular carcinoma. Dig Liver Dis. 2011 Jan;43 (4):333–4.
19.Couriel DR, Beguelin GZ, Giralt S, De Lima M, Hosing C, Kharfan-Dabaja MA. Chronic graft-versus-host disease manifesting as polymyositis: An uncommon presentation. Bone Marrow Transplant. 2002 Oct; 30 (8):543–6.
20.Dalakas MC. Inflammatory muscle diseases. N Engl J Med. 2015 Apr; 372 (18):1734–47.
21.Takahashi K, Kashihara K, Shinagawa K, Yoshino T, Abe K, Harada M. Myositis as a manifestation of chronic graft-versus-host disease. Intern Med. 2000 Jun; 39 (6):482–5.
22.Walsh M. Polymyositis [Internet]. PathologyOutlines.com. 2016. Available from: http://www.pathologyoutlines.com/topic/musclepolymyositis.html. Cited Mar 19, 2020
David Ben-Nun, 1 Medical Student, Sackler School of Medicine, Tel Aviv University, Tel Aviv, Israel
Mihnea-Alexandru Găman, Editor
Madeleine Jemima Cox, Student Editor
About the Author: David Ben-Nun is a fourth-year medical student at Sackler School of Medicine at Tel
Aviv University in Israel. He recently matched to the University of Texas at Austin
Dell Medical Center for a residency in internal medicine, which he will begin later
this year. He is currently involved in research on heart failure with preserved ejection
fraction, among other topics, and hopes to pursue a fellowship in cardiology following
residency.
Correspondence: David Ben-Nun, Address: 35, Tel Aviv-Yafo, 6997801, Israel Email: bennun@mail.tau.ac.il
Cite as: Ben-Nun D. Polymyositis as a Rare Musculoskeletal Manifestation of Chronic Graft-Versus-Host Disease: A Case Report of a 33-Year-Old Patient. Int J Med Students. 2020 Jan-Apr;8(1):36-40.
Copyright © 2020 David Ben-Nun
This work is licensed under a Creative Commons Attribution 4.0 International License.
International Journal of Medical Students, VOLUME 8, NUMBER 1, April 2020