Case Report
Familial Hyperinsulinism due to HNF4A Deficiency and Benign Premature Adrenarche:
A Case Report
Edward Compton1, David H. Geller2, Alaina P. Vidmar3
doi: http://dx.doi.org/10.5195/ijms.2021.721
Volume 9, Number 2: 167-170
Received 31 08 2020:
Rev-request 28 09 2020:
Rev-request 31 01 2021:
Rev-request 29 04 2021:
Rev-recd 22 10 2020:
Rev-recd 09 02 2021:
Rev-recd 02 05 2021:
Accepted 02 05 2021
ABSTRACT
Background:
Familial Hyperinsulinism due to HNF4A deficiency (FHI-HNF4A) is a form of diazoxide-sensitive,
diffuse hyperinsulinism, characterized by transient or persistent hyperinsulinemic
hypoglycemia, and a propensity to develop Maturity-Onset Diabetes of the Young type
1 (MODY1). The association between FHI-HNF4A deficiency and benign premature adrenarche
(BPA) is unknown.
The Case:
We report the case of a 5-year-old girl with FHI-HNF4A, controlled on diazoxide, who
presented with BPA and Tanner stage 3 pubic hair associated with body odor and acne.
Work-up revealed elevated dehydroepiandrosterone sulfate (DHEAS), elevated free testosterone,
and advanced bone age. Insulin levels were elevated in the setting of normal fasting
blood glucose. We discuss the possible hormonal underpinnings of hyperandrogenism.
Conclusion:
Though the underlying pathophysiology of this phenotype is unclear, a possible synergistic
mechanism exists between insulin-induced hyperandrogenism and HNF4A deficiency leading
to a transient decrease of SHBG and thus increased free testosterone levels. Further
investigation is required to determine the association between HNF4A dysfunction and
BPA.
Keywords:
Hyperinsulinism;
Congenital Hyperinsulinism;
Adrenarche;
HNF4A;
Hyperandrogenism (Source: MeSH-NLM).
Highlights:
- Familial Hyperinsulinism due to HNF4A deficiency (FHI-HNF4A) is a form of diazoxide-sensitive;
diffuse hyperinsulinism, characterized by transient or persistent hyperinsulinemic
hypoglycemia, and subsequent propensity to develop MODY1.
- By altering the HPA and HPG axes, hyperinsulinism may lead to increased levels of
circulating androgens, which has been demonstrated in conditions with insulin resistance
and subsequent hyperinsulinism, such as polycystic ovary syndrome (PCOS).
- Regulation of HNF4A by many factors indirectly regulates hepatic SHBG synthesis, as
HNF4A plays an imperative role in the regulation of SHBG.
- The association of elevated insulin levels, insulin resistance, and functional hyperandrogenism
has been previously described in youth with benign premature adrenarche (BPA) and
polycystic ovarian syndrome (PCOS) however there have been no reports in a patient
with FHI-HNF4A.
- These findings may suggest that patients with FFI-HNF4A may be at greater risk for
insulin induced hyperandrogenism and therefore diazoxide dosage should be titrated
to insulin levels to prevent functional hyperandrogenism and its sequelae.
- If HNF4A defects play a role in altering SHBG levels, it may be clinically relevant
to screen patients with BPA for these alterations.
Introduction
Congenital hyperinsulinism (CHI) is due to a variety of etiologies that result in
dysregulated insulin release from pancreatic β-cells. There are two histological variants
of CHI, focal and diffuse, which differ in the extent of pancreatic involvement. In
the diffuse variant, all of the β-cells are affected, while in the focal form, a localizable
lesion is found, affecting only a subset of the β-cells.1 Insulin secretion uncoupled from glucose metabolism results in hyperinsulinemic hypoglycemia.2,3 Familial Hyperinsulinism due to HNF4A deficiency (FHI-HNF4A) is a form of diazoxide-sensitive,
diffuse hyperinsulinism, characterized by macrosomia, transient or persistent hyperinsulinemic
hypoglycemia, and a propensity to develop Maturity-Onset Diabetes of the Young type
1 (MODY1). Patients with FHI-HNF4A are responsive to diazoxide treatment, which activates
KATP channels, leading to increased potassium conductance, cellular membrane hyperpolarization,
and inhibition of insulin release.4,5 Patients with CHI due to other genetic variants, in which the mutation lies within
subunits of the KATP channels themselves, are not responsive to diazoxide treatment.6 The role of HNF4A in FHI-HNF4A has yet to be fully elucidated, but it is thought
to work in combination with other transcription factors, forming a regulatory network
of proteins in the pancreatic islet.7 Mechanistically, HNF4A deficiency can impair the binding of HNF4A with p300, which
then prevents HNF4A from binding to the promoter region of HNF1. Interestingly, there
is a growing body of evidence that mRNA levels of HNF4A correlate with Sex Hormone
Binding Globulin (SHBG) mRNA levels. Therefore, HNF4A deficiency may result in decreased
SHBG, with subsequent increased levels of free testosterone.8 The clinical consequence of this process is poorly understood.
Benign premature adrenarche (BPA) is a clinical diagnosis often associated with elevations
of dehydroepiandrosterone (DHEA) and dehydroepiandrosterone sulfate (DHEAS) for chronological
age,.9,10 If phenotypic signs of androgen activity, such as pubic and/or axillary hair, adult-type
body odor, oily skin or hair, comedones, or accelerated growth velocity, are detected
before the age of 8 in females or 9 in increased rates of obesity in children with
BPA.9,10 However, other conditions that present similarly must first be excluded before BPA
can be diagnosed. These include: central puberty, adrenocortical and gonadal sex-hormone
secreting tumors, congenital adrenal hyperplasia, and exposure to exogenous androgens.
In some populations, BPA has been associated with low birth weight, insulin resistance,
adverse cardiometabolic risk, and progression to polycystic ovary syndrome (PCOS).11–15 Herein, we report a 5-year-old female patient with FHI-HNF4A who presented with BPA
in the setting of elevated insulin level, despite euglycemia on diazoxide therapy.
The underlying pathophysiology of this phenotype remains obscure; however, we discuss
a possible synergistic mechanism between insulin-induced hyperandrogenism and HNF4A
deficiency, leading to transient decrease of SHBG and thus increased free testosterone
levels.
The Case
A 5-year-old girl with known FHI-HNF4A, who was first noted by her mother to have
new-onset acne and body odor without any associated breast changes, pubic hair, or
menses at 4 years of age, presented to the pediatric endocrinology clinic for follow-up.
Her pubic hair had progressed to Tanner stage 3 with increased acne and body odor.
No exogenous steroid exposures were reported. Physical exam revealed Tanner stage
1 breasts. Laboratory testing revealed abnormalities in the following: free testosterone
0.6 ng/dL (normal range < 0.04-0.14 ng/dL), and DHEAS 146 mcg/dL (normal range: ≤
34 mcg/dL). The following were within reference ranges: fasting laboratory testing
revealed a HbA1c of 4.0% (normal range: 3.0-5.8%), a fasting blood glucose of 80 mg/dL
(normal range: 70-100 mg/dL), anti-Mullerian hormone 1.3 ng/mL, estradiol < 7 pg/mL
(normal range: < 7 pg/mL), total testosterone < 7 ng/dL (normal range: <7-20 ng/dL),
insulin 11.3 uIU/mL (normal range: 2.0-19.6 uIU/mL), 17-hydroxypregnenolone 177 ng/dL
(normal range: ≤ 561 ng/dL), 17-hydoxyprogesterone 35 ng/dL (normal range: ≤ 137 ng/dL),
serum androstenedione 18 ng/dL (normal range : ≤ 45 ng/dL and serum DHEA 159 ng/dL
(normal range: ≤ 487 ng/dL) (Table 1). A bone age study revealed an advanced bone age of 7.8 years and at this time, the
decision was made to monitor the patient serially, without altering diazoxide dosing.
Table 1.
Clinical and laboratory findings at most recent visit.
| Characteristics |
Patient Values |
Reference Range (if applicable) |
| Age, years |
6.0 |
|
| Bone age, years |
7.8 |
|
| Weight, kg (%ile) |
17.3 (15) |
|
| Height (%ile) |
110.7 (27) |
|
| BMI (%ile) |
14.12 (18) |
|
| Tanner Stage (breast/pubic hair) |
1/3 |
|
| Glucose, mg/dL |
80 |
60–115 |
| HbA1c |
4.3 |
3.0–6.0 |
| Anti-Mullerian Hormone |
1.3 |
|
| Estradiol, pg/mL |
< 7 |
<7 |
| Total Testosterone, ng/dL |
< 7 |
< 7 |
| Insulin, uIU/mL |
11.3 |
2.0–19.6 |
| 17-hydroxyprogesterone, ng/dL |
35 |
≤ 137 |
| 17-hydroxypregnenolone, ng/dL |
177 |
≤ 561 |
| Serum androstenedione, ng/dL |
18 |
≤ 45 |
| DHEA sulfate, mcg/dL |
146 |
≤ 34 |
| DHEA, ng/dL |
159 |
≤ 487 |
Legend:
BMI = body mass index; DHEA = Dehydroepiandrosterone
The patient's past medical history was significant for premature delivery at 34 weeks
gestation due to premature rupture of membranes. The vaginal delivery was complicated
by prolonged labor and fetal distress. Her birth weight was 1870 grams, with an associated
length of 43.15 cm, appropriate for gestational age. She had a macrosomic appearance
at birth. She was admitted to the neonatal intensive care unit for fifty days, requiring
supplemental oxygen for twelve days. She was found to have persistent hypoglycemia,
requiring a glucose infusion rate of 18-20 mg/kg/minute. She was diagnosed with hyperinsulinemic
hypoglycemia on day five of life and was started on diazoxide at 15 mg/kg/day. Subsequent
laboratory and genetic testing confirmed her heterozygous HNF4A mutation (NM_00457.4
c.253C>T, p.Arg85trp – PubMed. 2030154, OMIM. 6160266), pathogenic for FHI-HNF4A.
By the age of 2, she was taking 100% of her caloric needs orally, requiring gastrostomy
tube (G-tube) supplementation only during times of illness. Her blood glucose levels
were within the target range 90% of the day based on continuous glucose monitoring
data and her HbA1c was 4.5%. Her father was diagnosed with FHI-HNF4A in infancy; at
the age of 20, was found to have hyperglycemia consistent with MODY1.
The family was lost to follow up and presented for referral to the pediatric endocrinology
clinic for consultation and management of congenital hyperinsulinism when she was
2.5 years old. At this time, she weighed 10.5 kg (< 3rd percentile), with a height of 87.5 cm (10th percentile) and BMI of 13.71 kg/m2 (< 3rd percentile). On physical exam, she had numerous dysmorphic features including macrosomia,
down-turned palpebral fissures, broad midface, low-set ears, and broad, wide-set thumbs.
She had diffuse hypertrichosis of the arms, face, back, and abdomen. Her external
genitalia were normal for age, with Tanner stage 1 axillary and pubic hair, and breasts
with Tanner stage 1 development. At this time, blood glucose levels were being monitored
only once daily, and it was recommended that blood glucose checks be increased to
6 times daily to ensure that cryptic hypoglycemia was detected, as HbA1c was < 4.0%.
She was placed on an iPro glucose monitor (CGM) to collect continuous glucose levels
for 96 hours, which demonstrated hypoglycemia 20% of the time. She was maintained
on diazoxide at 12 mg/kg/day divided three times daily (TID). Due to concern for poor
annualized growth velocity of 1.4 cm/year, a bone age was obtained, which showed skeletal
age concordant with chronological age.
At a subsequent follow-up at 3.5 years old, her HbA1c was 4.0%, with weight gain and
associated increased annualized growth velocity of 6 cm/yr. Continuous glucose monitoring
was recommended to detect overnight hypoglycemic episodes, but the patient's family
declined this option. She was receiving feeding therapy and her oral intake had improved
significantly. Although she no longer utilized G-tube feedings, she was having multiple
episodes of hypoglycemia overnight. Her diazoxide dose was increased to 13 mg/kg/day
divided TID, with resolution of her overnight hypoglycemia.
Discussion
We report a unique case of a 5-year-old female with FHI-HNF4A who presented with Tanner
3 pubic hair, acne, body odor, elevated DHEAS and free testosterone, and advanced
bone age in the absence of elevated estradiol levels. Her presentation is most consistent
with BPA; however, the relationship between FHI-HNF4A and BPA remains poorly understood.
Although the BMI percentile of our patient was 18%, prior studies have found an association
between BPA and obesity.9,10 There is a wide differential diagnosis for patients that are found to be persistently
hypoglycemic after birth, including hyperinsulinism as in our patient, mutations in
enzymes involved in fatty acid metabolism, glycogen storage disorders, counter-regulatory
hormone deficiencies. While the differential is broad, we suspected hyperinsulinemia
as the culprit in our patient, given her father's diagnosis of FHI in infancy. Thus,
genetic testing was obtained early in her disease course. Many of the etiologies of
persistent hypoglycemia in infancy involve a genetic component, highlighting the importance
of obtaining a satisfactory family history, as this can help narrow the differential
diagnosis, expediting achieving a diagnosis.
Besides HNF4A, other genes have been implicated in hyperinsulinemia, with the two
most common being mutations in ABCC8 and KCNJ11. Mutations in these two genetic loci
are responsible for both focal and diffuse forms of CHI, leading to vastly different
treatments and outcomes. Therefore, genetic analysis becomes important for counseling
families on treatment and prognosis, as one study demonstrated that detecting a single
paternally derived mutation predicted focal disease 94% of the time.16
One possible mechanism to explain the observed association is that our patient was
experiencing transient hyperinsulinism resulting in increased adrenal androgen production.
Insulin is hypothesized to play a role in the regulation of the hypothalamic-pituitary-adrenal
(HPA) and hypothalamic-pituitary-gonadal (HPG) axes.17,18 A variety of regulatory mechanisms have been proposed, including altered expression
of key enzymes involved in steroidogenesis, increased secretion of gonadotropin-releasing
hormone (GnRH), increased amplitude of luteinizing hormone (LH) pulses, potentiation
of ACTH-stimulated steroidogenesis, and inhibition of SHBG production.8 By up-regulating one or both of the HPA and HPG axes, hyperinsulinism may lead to
increased levels of circulating androgens. In patients with conditions such as PCOS,
which have been associated with BPA, elevated total and free androgens have been demonstrated
in the setting of insulin resistance and decreased SHBG.19,20 However, prior studies have not explored the potential relationship between FHI-HNF4A
or other genetic CHI disorders, and BPA. Mechanistically, insulin has been demonstrated
to increase mRNA levels of CYP17A1 and 3?-HSD and potentiate ACTH production of intermediates
involved in DHEAS synthesis, leading authors to conclude that the hyperandrogenic
features observed in PCOS may be due in part to a hyperinsulinemia-derived increase
in adrenal androgens.21,22
Ibanez et al. studied 10 non-obese adolescent females who had experienced BPA, hirsutism, ovarian
hyperandrogenism, oligomenorrhea, dyslipidemia, and hyperinsulinemia. These females
were administered metformin daily for 6 months, which reduced insulin levels, hirsutism,
and hyperandrogenism, and attenuated the release of LH in response to GnRH pulses.
One of the mechanisms by which metformin improves hyperglycemia in diabetes is through
the inhibition of mitochondrial glycerophosphate dehydrogenase, which inhibits gluconeogenesis
and increases insulin sensitivity, thereby decreasing insulin requirements. Metformin
administration also increases SHBG, leading to decreased serum-free androgens.23,24 Importantly, the authors noted that DHEAS decreased with metformin, buttressing the
argument that insulin increased adrenal androgen production.23 In a study contrasting 47 adolescent females with BPA vs 22 healthy female controls,
Kaya et al. found that females with BPA had higher body mass indexes and insulin concentrations.10 These females also had hyper-responsiveness to ACTH, leading to increased androstenedione
and DHEA levels. This study further elucidates the unique role insulin plays in the
regulation of adrenal sex hormone production.
An additional mechanism to consider in this patient is the effect of HNF4A function
on SHBG levels. Hammond et al. proposed that activation of the SHBG promoter in the liver involves HFN4 binding
to a DR1-like cis-element which then stimulates production.25 Therefore, in HNF4A deficiency, SHBG production would be decreased, with consequent
increases in free androgen levels. Winters et al. found a strong positive correlation between the level of HNF4A mRNA and SHBG mRNA,
with an inverse relationship between insulin resistance/insulin levels, and circulating
SHBG and HNF4A mRNA levels.26 These findings suggest that circulating SHBG levels may be mediated by HNF4A and
provide further insight into the mechanism by which HNF4A deficiency could predispose
a patient with FHI-HNF4A to BPA. The combination of these two molecular entities could
have resulted in the BPA-like phenotype observed in our patient. A summary of the
proposed mechanisms of insulin regulation of androgen synthesis and circulatory levels
is shown in Figure 1.
Figure 1
Proposed mechanisms of insulin regulation of androgen synthesis and circulatory levels.
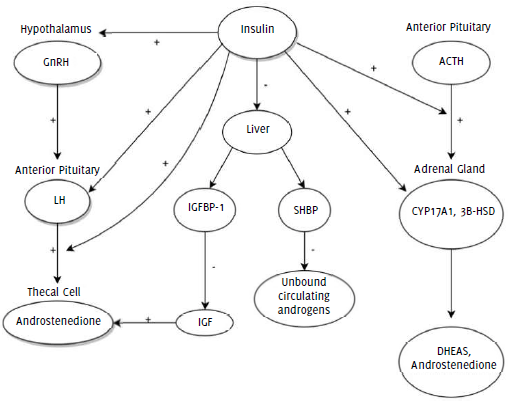
Legend: 3B-HSD = 3β-hydroxysteroid dehydrogenase; ACTH = Adrenocorticotropic hormone; CYP17A1
= cytochrome P450 17A1; DHEAS = dehydroepiandrosterone sulfate; GnRH = gonadotropin
releasing hormone; IGF = insulin-like growth factor; IGFBP-1 = insulin-like growth
factor binding protein 1; LH = luteinizing hormone; SHBP = sex hormone binding protein.
This case demonstrates that clinically silent hypoglycemia with concomitant intermittent
hyperinsulinemia may have long-term sequelae for the patient. Therefore, even if glycemic
control is adequate overall, with HbA1c levels within normal limits, it is important
not to ignore either glucose levels or HbA1c levels that are down-trending. Large
fluctuations in blood glucose levels should be avoided, with close monitoring of daily
glucose checks. In our patient, glucose checks were only being performed once daily;
subsequent continuous glucose monitoring demonstrated hypoglycemia 20% of the time.
This highlights the importance of more rigorous glucose monitoring in these patients,
as they may not present with the normal signs of hypoglycemia (diaphoresis, lightheadedness,
tachycardia, etc.) and thus their hypoglycemia may go clinically undetected until
more severe sequela develops. This has important treatment implications, as tight
medical management becomes paramount. While our patient responded well and tolerated
diazoxide, other patients may not respond as well, requiring additional treatment
considerations to adequately control glucose, and subsequently, insulin levels.
We believe that our patient may have been experiencing episodes of hypoglycemia, as
evidenced by her HbA1C levels on the low end of normal. Episodes of hypoglycemia may
have been due to intermittent hyperinsulinemia in spite of diazoxide treatment. We
hypothesize these periods of hyperinsulinemia may have been sufficient to increase
adrenal steroidogenic activity, and subsequently, increased circulating levels of
DHEAS. Furthermore, her HNF4A deficiency may have led to a decrease in SHBG levels
resulting in elevated free testosterone. Future studies are needed to 1) investigate
the underlying molecular etiology of hyperandrogenism in patients with FHI-HNF4A and
BPA, 2) elucidate the optimal dosage of diazoxide treatment in FHI-HNF4A to prevent
any long-term sequelae that could occur in the setting of transient hyperinsulinemia,
and 3) explore the relationship between HNF4A deficiency and BPA.
Conflict of Interest Statement & Funding
The Authors have no funding, financial relationships or conflicts of interest to disclose.
Author Contributions
Conceptualization: DG, AV. Data Curation: EC, AV. Formal Analysis, Methodology, Visualization,
& Writing – Review and Editing: EC, DG, AV. Investigation & Project Administration:
EC, AV. Supervision: AV. Validation: DG, AV. Writing – Original Draft: EC.
Acknowledgments
None.
References
1. Lord K, Dzata E, Snider KE, Gallagher PR, De Leon DD. Clinical Presentation and Management of Children with Diffuse Hyperinsulinism: A Review
of 223 Cases. J Clin Endocrinol Metab. 2013; 98(11): E1786–E1789.
2. Ferrara C, Patel P, Becker S, Stanley CA, Kelly A. Biomarkers of Insulin for the Diagnosis of Hyperinsulinemic Hypoglycemia in Infants
and Children. J Pediatr. 2016;168:212–9.
3. Aynsley-Green A, Hussain K, Hall J, et al. Practical management of hyperinsulinism in infancy. Arch Dis Child Fetal Neonatal Ed. 2000;82(2):F98–F107.
4. Flanagan SE, Kapoor RR, Mali G, et al. Diazoxide-responsive hyperinsulinemic hypoglycemia caused by HNF4A gene mutations. Eur J Endocrinol. 2010;162(5):987–92.
5. Kapoor RR, Locke J, Colclough K, et al. Persistent hyperinsulinemic hypoglycemia and maturity-onset diabetes of the young
due to heterozygous HNF4A mutations. Diabetes. 2008;57(6):1659–63.
6. Lord K, De Leon DD. Monogenic hyperinsulinemic hypoglycemia: current insights into the pathogenesis and
management. Int J Pediatr Endocrinol. 2013;3.
7. Odom DT, Zizlsperger N, Gordon DB, et al. Control of pancreas and liver gene expression by HNF transcription factors. Science. 2004;303(5662):1378–81.
8. Winters SJ, Gogineni J, Karegar M, et al. Sex hormone-binding globulin gene expression and insulin resistance. J Clin Endocrinol Metab. 2014;99(12):E2780–2788.
9. Utriainen P, Laakso S, Liimatta J, et al. Premature adrenarche–a common condition with variable presentation. Horm Res Paediatr. 2015;83(4):221–31.
10. Kaya G, Yavas Abali Z, et al. Body mass index at the presentation of premature adrenarche is associated with components
of metabolic syndrome at puberty. Eur J Pediatr. 2018 Nov;177(11):1593–601.
11. Ibáñez L, Potau N, Francois I, et al. Precocious pubarche, hyperinsulinism, and ovarian hyperandrogenism in girls: relation
to reduced fetal growth. J Clin Endocrinol Metab. 1998 Oct;83(10):3558–62.
12. Ehrmann DA, Barnes RB, Rosenfield RL. Polycystic ovary syndrome as a form of functional ovarian hyperandrogenism due to
dysregulation of androgen secretion. Endocr Rev. 1995 Jun;16(3):322–53.
13. Ibáñez L, Potau N, Zampolli M, et al. Hyperinsulinemia and decreased insulin-like growth factor-binding protein-1 are common
features in prepubertal and pubertal girls with a history of premature pubarche. J Clin Endocrinol Metab. 1997 Jul;82(7):2283–8.
14. Ibáñez L, Potau N, Chacon P, et al. Hyperinsulinaemia, dyslipaemia and cardiovascular risk in girls with a history of
premature pubarche. Diabetologia. 1998 Sep;41(9):1057–63.
15. Després JP, Lamarche B, Mauriège P, et al. Hyperinsulinemia as an independent risk factor for ischemic heart disease. N Engl J Med. 1996 Apr 11;334(15):952–7.
16. Snider KE, Becker S, Boyajian L, et al. Genotype and phenotype correlations in 417 children with congenital hyperinsulinism. J Clin Endocrinol Metab. 2013 Feb;98(2):E355–63.
17. Fruehwald-Schultes B, Kern W, Bong W, et al. Supraphysiological hyperinsulinemia acutely increases hypothalamic-pituitary-adrenal
secretory activity in humans. J Clin Endocrinol Metab. 1999 Sep;84(9):3041–6.
18. Adashi EY, Hsueh AJ, Yen SS. Insulin enhancement of luteinizing hormone and follicle-stimulating hormone release
by cultured pituitary cells. Endocrinology. 1981 Apr;108(4):1441–9.
19. Nestler JE, Powers LP, Matt DW, et al. A direct effect of hyperinsulinemia on serum sex hormone-binding globulin levels in
obese women with the polycystic ovary syndrome. J Clin Endocrinol Metab. 1991 Jan;72(1):83–9.
20. Nestler JE, Barlascini CO, Matt DW, Steingold KA, Plymate SR, Clore JN, Blackard WG. Suppression of serum insulin by diazoxide reduces serum testosterone levels in obese
women with polycystic ovary syndrome. J Clin Endocrinol Metab. 1989 Jun;68(6):1027–32.
21. Rosenfield RL. Evidence that idiopathic functional adrenal hyperandrogenism is caused by dysregulation
of adrenal steroidogenesis and that hyperinsulinemia may be involved. J Clin Endocrinol Metab. 1996;81(3):878–80.
22. Nestler JE, Jakubowicz DJ. Decreases in ovarian cytochrome P450c17 alpha activity and serum free testosterone
after reduction of insulin secretion in polycystic ovary syndrome. N Engl J Med. 1996;335(9):617–23.
23. Ibanez L, Valls C, Potau N, et al. Sensitization to insulin in adolescent girls to normalize hirsutism, hyperandrogenism,
oligomenorrhea, dyslipidemia, and hyperinsulinism after precocious pubarche. J Clin Endocrinol Metab. 2000;85(10):3526–30.
24. la Marca A, Morgante G, Paglia T, et al. Effects of metformin on adrenal steroidogenesis in women with polycystic ovary syndrome. Fertil Steril. 1999;72(6):985–9.
25. Hammond GL. Diverse roles for sex hormone-binding globulin in reproduction. Biol Reprod. 2011;85(3):431–41.
26. Winters SJ, Gogineni J, Karegar M, et al. Sex hormone-binding globulin gene expression and insulin resistance. J Clin Endocrinol Metab. 2014 Dec;99(12):E2780–8.
Edward Compton, 1 BS, The Center for Endocrinology, Diabetes and Metabolism, Children's Hospital Los
Angeles, Los Angeles, CA, United States
David H. Geller, 2 MD, PhD, The Center for Endocrinology, Diabetes and Metabolism, Children's Hospital
Los Angeles, Los Angeles, CA, United States
Alaina P. Vidmar, 3 MD, The Center for Endocrinology, Diabetes and Metabolism, Children's Hospital Los
Angeles, Los Angeles, CA, United States
About the Author: Edward Compton is currently a 3rd year medical student at Keck School of Medicine, Los Angeles, CA, USA of a 4-year
program.
Correspondence: Edward Compton, Address: 4650 Sunset Blvd, Los Angeles, CA 90027, United States.
Email: ecompton@usc.edu
Editor: Francisco J. Bonilla-Escobar
Student Editors: Azher Syed
Student Editors: Brandon Belbeck
Student Editors: Bahadar Srichawla
Copyeditor: Benjamin Liu
Proofreader: Joseph Tonge
Layout Editor: Annora Ai-Wei Kumar
Cite as:
Compton E, Geller DH, Vidmar AP. al. Familial Hyperinsulinism due to HNF4A Deficiency
and Benign Premature Adrenarche: A Case Report. Int J Med Students. 2021 May-Jun;9(2):167-70.
Copyright © 2021 Edward Compton, David H. Geller, Alaina P. Vidmar MD
This work is licensed under a Creative Commons Attribution 4.0 International License.
International Journal of Medical Students, VOLUME 9, NUMBER 2, May-June 2021