The Amyloid Hypothesis: Amyloidogenic and Non-amyloidogenic Pathways.
Evelyn Chou1
doi: http://dx.doi.org/10.5195/ijms.2014.85
Volume 2, Number 2: 56-63
Received 15 10 2013: Accepted 14 05 2014
ABSTRACT
Alzheimer's disease (AD) is a currently incurable neurodegenerative disorder whose treatment poses a big challenge. Proposed causes of AD include the cholinergic, amyloid and tau hypotheses. Current therapeutic treatments have been aimed at dealing with the neurotransmitter imbalance. These include cholinesterase inhibitors and N-Methyl-D-aspartate (NMDA) antagonists. However, current therapeutics have been unable to halt AD progression. Much research has gone into the development of disease-modifying drugs to interfere with the course of the disease. Approaches include secretase inhibition and immunotherapy aimed at reducing plaque deposition. However, these have not been successful in curing AD as yet. It is believed that the main reason why therapeutics have failed to work is that treatment begins too late in the course of the disease. The future of AD treatment thus appears to lie with prevention rather than cure. In this article, current therapeutics and, from there, the future of AD treatment are discussed.
Keywords: Alzheimer's disease; disease-modifying drugs; beta-secretase inhibitors; gamma-secretase inhibitors; cholinergic; amyloid; tau.
The most common form of dementia is Alzheimer's disease (AD). It is a degenerative and currently incurable terminal disease, affecting about 75% of the 35 million people worldwide suffering from dementia. It is predicted that the prevalence of AD will double every 20 years, meaning an estimated 115 million individuals may be suffering from AD by 2050.1 AD is thus becoming increasingly recognized as a major cause of medical and social burdens in the elderly population worldwide.2 In its preclinical stages, AD cannot be diagnosed, while its clinical stages are characterized by impairment of cognitive functions (i.e. recent memory, language difficulties, spatial disorientation and visual agnosia), with behavioural disturbances significant enough to compromise activities of daily living (ADLs). Life expectancy is reduced, with patients generally living up to 5-8 years following diagnosis.3
Currently, no drugs are available to halt the progression of neurodegeneration in AD; the nature of AD treatment is symptomatic.2 For instance, cholinesterase inhibitors (CIs) that promote cholinergic neurotransmission are used in mild to moderate cases of AD. Memantine, an N-methyl-D-aspartate (NMDA) receptor antagonist, is used in moderate to severe cases to prevent excitotoxicity,4 and antipsychotics and antidepressants are used in the treatment of neuropsychiatric symptoms.5
The future of treatment of AD lies in the targeting of neuritic plaques (NPs) and neurofibrillary tangles (NFTs), which has the potential to delay neurodegeneration.6
The daunting statistics and the impacts that AD has on sufferers, caregivers and the society make it exceptionally vital that we review how AD is currently being treated and how this is likely to change in the near future with the possible development of disease modifying treatments.
The papers for this review article were identified by computerized advanced searches in Pubmed database and Google Scholar using the keywords ‘Alzheimer's’, ‘beta-secretase’ and‘gamma-secretase’. These papers included meta-analyses, original research articles, review articles and clinical trials. Information was also obtained from textbooks and Alzheimer's disease forums. This review follows the Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) Statement.7
The progression of AD can be divided into a series of stages: pre-dementia, mild, moderate and severe.
The pre-dementia stage is often unreliably distinguished from normal aging or stress-related issues.3,8 One of the first signs is the deterioration of episodic memory. No decline in sensory or motor performance occurs at this stage, and other aspects such as executive, verbal and visuospatial functions are slightly impaired at most. An individual remains independent and is not diagnosed as suffering from AD.8
During mild stages of AD, increased memory loss affects recent declarative memory more profoundly than other capacities, such as short-term, declarative and implicit memories.3
Motor function remains normal, and sensory performance is not impaired extensively. However, some visual, auditory and olfactory functions may be affected.8 Communication begins to decline as patients find themselves unable to recall certain words. The individual may be able to remain independent, but not without some assistance.3,9
Recent memory continues to deteriorate in the moderate stage. Due to an inability to create new memories, AD patients seem to live in the past.9 Patients are still able to manage basic ADLs, but help is required in certain areas such as grooming and dressing.3,9 Insight into their disease is commonly lost by this stage, with patients becoming delusional. A longitudinal study conducted in 1993 showed that it is at this stage that cognitive decline, aggression, depression and incontinence in patients become predictive factors for placement in nursing homes.10
In the severe stage, even early memories can be lost. Basic ADLs are now affected, declining gradually. Communication deteriorates further to single words or phrases, and language is thus significantly impaired.3,9 Behavioural disturbances occur, causing disruptions to caregivers.3,11 The most common cause of death in AD patients is pneumonia,12 followed by myocardial infarction (MI) and septicaemia.3
Inheritance of certain genes is a risk factor for AD, with both familial and sporadic cases occurring. In sporadic AD, which is the more common form, there is a link with the apolipoprotein ε04 (APOE4) allele, with the risk being greater in homozygotic situations.1,13 It has been shown that transgenic mice expressing either mouse or human apoE develop neuritic plaques (NPs) associated with neuritic degeneration due to fibrillar amyloid-ß deposits. n contrast, in apoE negative mice, no neuritic degeneration was observed despite the presence of non-fibrillary amyloid-ß deposits. ApoE thus appears to play a critical role in the progression of NPs and degeneration.14
Environmental factors also contribute to the development of sporadic AD.15 Familial AD, on the other hand, has been associated with mutations in presenilin 1 (PSEN1) and presenilin 2 (PSEN2), as well as the amyloid precursor protein (APP) gene, which is located on chromosome 21.1,13 Many other candidate polymorphic genes have been associated with increased AD risks, including secretase, peptidase, microtubule, cytoskeletal, anti-apoptotic and protease genes.1
Vascular factors seem to affect the risks of developing AD. Metabolic syndrome,1 comprising hypertension, dyslipidaemia, obesity and diabetes mellitus, has been associated with increased risk.16 Hypertension contributes to the formation of neuritic plaques (NPs), neurofibrillary tangles (NFTs), and characteristic lesions seen in AD.17 Dyslipidaemia and diabetes mellitus are not only implicated in the generation of NPs, but also cause cerebrovascular dysfunction.18 In addition, obesity has been linked to cognitive decline and resistance to insulin the latter of which leads to hypertension, diabetes and cerebrovascular dysfunction.19
Psychosocial factors are also implicated in AD. With greater social, physical and mental stimulation, individuals are less likely to develop AD later in life. The risk of cognitive decline is higher in those with lower physical activity. Additionally, the protective effect of physical activity is more prominent in apoE4 allele carriers. These three lifestyle factors appear to act together in a common pathway in their protection against dementia.1,20
The cholinergic hypothesis of AD came about due to the combined observations of deficits in choline acetyltransferase and acetylcholine (ACh) and the fact that ACh is important in memory and learning. It was thought that reduction in cholinergic neurons as well as cholinergic neurotransmission led to the decline in cognitive and noncognitive functions. Cholinergic function loss correlated to cognitive decline, but no causal relationship was established.2,21 Moreover, the use of cholinesterase inhibitors (CIs) does not have a significant effect in more than half of AD patients receiving treatment, indicating the presence of other important processes in the progression of the disease.21
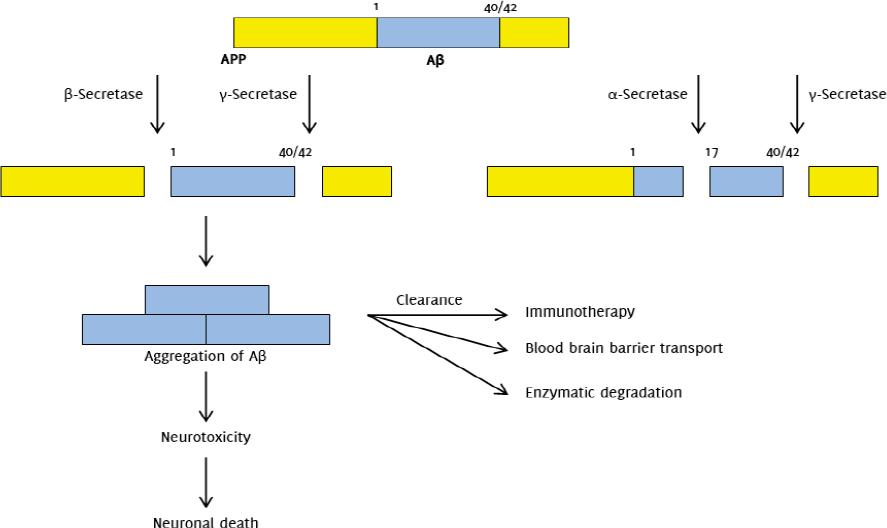
Amyloidosis is the abnormal deposition of amyloid proteins in tissues, with the altered amyloid proteins forming an insoluble ß-pleated sheet. Reduced tissue and cellular clearance is observed in amyloid protein deposits. The membrane protein amyloid-ß precursor protein (APP) is proteolysed to form Aß, and it is the amyloid form of Aß that makes up the amyloid plaques (neuritic plaques) found in the brains of AD sufferers.6
According to the amyloid hypothesis, the basis of AD is the presence of Aß production in the brain.2 Evidence for the amyloid hypothesis was compelling, as gene mutations encoding the amyloid-ß precursor protein (APP) was found to cause familial AD, with sites of major mutations found in γ secretase and APP.6 Aß is derived from APP by proteolysis in the amyloidogenic pathway, mediated by ß secretase (BACE1) and γ secretase, in the extracellular and transmembrane region, respectively. Cleavage by ß-secretase produces APPsß and C99. C99 is further cleaved by γ secretase to form either Aß1-40 or the more hydrophobic, aggregation-prone Aß1-42.22
Aß40 is more predominant in cerebral vasculature.2 APP can also be cleaved by α secretase in the non-amyloidogenic pathway, producing APP α and C83. Further evidence came from an experiment in the 1990s whereby transgenic mice expressing three different isoforms of mutant APP were found to have characteristic AD neuropathologies.23
Despite widespread support of Aß fibrils being the main cause of pathology seen in AD, it was suggested that oligomerization of Aß1-42 plays a more important role. Oligomerization of Aß1-42 produces soluble Aß oligomers which are known as Aß-derived diffusible ligands (ADDLs). Experiments showed that these ADDLs are potentially more toxic than Aß fibrils as they target synaptic spines and disrupt synaptic plasticity, thus affecting cognitive function. Their toxicity lies in toxin receptors on cell surfaces and in Fyn, a tyrosine kinase receptor overexpressed in AD (Figure 1).24,25
Figure 1.The Amyloid Hypothesis: Amyloidogenic and Non-amyloidogenic Pathways.
The Tau hypothesis revolves around the presence of neurofibrillary tangles (NFTs) in AD. As a result of increased phosphorylation of Tau (originally bound to microtubules), there is an increase in free tau accompanied by loss of functioning microtubules.26 Phosphorylated Tau are subunits of paired helical filaments (PHFs), which form NFTs. The impaired microtubules affect axonal transport of proteins and eventually cause neuronal death.27
The degeneration of neurons and synapses, declining cholinergic function, characteristic neuropathologic lesions of neuritic plaques (NPs) and neurofibrillary tangles (NFTs) all contribute to cognitive decline.28 The neocortex and hippocampus, which are the regions of higher function, are most affected by these lesions.21 NPs consist of aggregations of amyloid-ß peptide, while NFTs are located within neurons and their projections and are composed of filamentous hyperphosphorylated tau. The distinction between NPs and NFTs being causative or mere markers of AD and the chronological order in which the pathologies appear are important to the understanding of AD pathology.6 A study carried out to determine the role of NPs and NFTs in AD established that NP deposition occurs in early stages of the disease, but does not correlate with progression of illness once clinical stages have been established, whereas NFTs seem to correlate to decline in cognitive function in later stages.28
It is through the understanding of the disease processes that underlie AD that targets can be ascertained and treatments developed.
Cholinesterase inhibitors (CI) aim to increase acetylcholine availability in synaptic neurotransmission in order to treat memory disturbances. Currently, three CIs are being used as the first-line treatment in mild to moderate AD: donepezil, rivastig-mine and galantamine.2 While donepezil and rivastigmine are both selective inhibitors, galantamine inhibits both ACh and butyrylcholinesterase. A meta-analysis collaborating 13 randomized, double-blind trials that were designed to evaluate the effectiveness and safety of CIs showed no improvement in ADL and behaviour. In addition, donepezil and rivastigmine showed no significant difference in their impact on cognitive functions, ADLs and behaviour. Overall, similar benefits were observed across all three drugs.29 It is known that CIs are unable to halt disease progression, but they have been found to have effects for a substantial period of time. As seen in a randomized double-blind trial, patients undergoing long-term treatment with donepezil showed no beneficial loss for up to two years.30
In addition, there may be some added benefits to increased doses of CIs given. In a randomized, double-blind, parallel-group, 48-week study conducted to determine the efficacy and safety of a rivastigmine patch of a higher dose, deterioration of ADLs was significantly reduced and Alzheimer's Disease Assessment Scale-cognitive subscale (ADAS-cog) was improved in patients treated with higher doses.31 Side effects as a result of CIs are minimal and are usually limited to gastrointestinal symptoms such as diarrhea, nausea and vomiting.13
Memantine is a non-competitive NMDA receptor antagonist effective in the treatment of moderate-to-severe AD. The modulation of NMDA receptors results in reduced glutamate-induced excitotoxicity. Its benefits were proven in a 28-week, double blind, parallel-group study which showed that treatment significantly reduced deterioration in patients. Most adverse reactions to the drug were not severe and were considered to be unrelated to the drug. The positive effect on cognitive function translates to behavioural improvements: patients were less agitated and required less assistance from caregivers.32 Improvement of the behavioural and psychological symptoms related to dementia (BPSD) was also highlighted by a meta-analysis of 6 studies involving memantine treatment.33
BPSD is a common occurrence in AD and a major source of burden on caregivers. CIs and memantine help to control these symptoms to a certain extent, but as patients continue to deteriorate, control by these drugs becomes insufficient.2
Depression is very common, especially in the early and late courses of the disease. Antidepressants such as: selective serotonin reuptake inhibitors (SSRI: citalopram, fluoxetine, paroxetine, sertraline, trazodone), tricyclic agents and combined serotonergic and noradrenergic inhibitors may be used to counter this.2,34 Discontinuation of antidepressants in demented patients in a double blinded, randomized, parallel-group placebo controlled trial showed significant increases in depression when compared to those who continued treatment. These results are indicative of the beneficial effects of antidepressants.34
Atypical antipsychotics used in AD include olanzapine, quetia-pine and risperidone, which are used to treat psychosis and agitation. However, the use of such drugs appears to be controversial, with patients showing significant declines in cognitive function with antipsychotic drugs administration when compared to patients receiving the placebo.35
While symptomatic treatments have proven helpful, it is the finding of a cure that is most vital. Since the amyloid hypothesis indicates that Aß generation and deposition from overexpressed APP cleavage make up the fundamental basis of AD, interest centers on anti-amyloid therapies. These therapies result in decreased production of Aß, increased clearance of Aß and the prevention of Aß aggregation into amyloid plaques.6,36 Immunotherapy has also been an area of interest as it targets the clearing of Aß peptides, which can either directly or indirectly impact cognitive decline.37
Focusing on decreasing Aß generation, several methods can be employed to achieve this, mainly by targeting the amyloidogenic and nonamyloidogenic pathways. ß and α. secretases both compete for APP, with ß- and γ-secretase processing ultimately resulting in amyloid deposition and γ-secretase generating soluble APPsα.2 Inhibiting ß- and γ-secretases while simultaneously potentiating α-secretase action would thus reduce Aß generation and deposition overall.
The cloning of ß-secretase has allowed for the investigation of its structural and catalytic properties.38 Development of clinically effective inhibitors depends greatly on this detailed knowledge of its structure. ß-secretase is a rate-limiting transmembrane aspartyl protease beta-site APP-cleaving enzyme (BACE1) involved in the first proteolytic step of the amyloidogenic pathway of Aß generation. BACE1-deficient mice were used to determine the role of ß-secretase. In mice with BACE1-/- neurons, generation of Aß40 and 42 was eradicated, thus confirming the role of BACE1 in the amyloidogenic pathway.39 From this, it can be inferred that inhibition of BACE1 activity would be able to reduce Aß levels and ultimately reduce neuritic plaques. Thus, it has become a primary therapeutic target.
The close resemblance of ß-secretase's catalytic apparatus to other aspartyl protease targets such as HIV protease has contributed to the development of its inhibitors.39 However, the secretase's hydrophilic, long and shallow active site presents some problems to the development of its inhibitors. An effective inhibitor would need to be able to penetrate the blood-brain-barrier (BBB) and the cell membrane of neurons. This has made the discovery of small yet potent inhibitors more difficult.36 Another challenge is that ß-secretase inhibitors act as substrates for P-glycoprotein that transports them out of the brain. Efforts have been made to overcome this through the design of inhibitors with higher selectivity for BACE1 over memapsin 1 (BACE2) and cathepsin D (cathD).2,39 The low number of drugs reaching clinical trials reflects the difficulty of these challenges faced (Figure 2).
Figure 2.Possible Therapeutic Targets in Anti-amyloid Therapies.
The earliest BACE1 inhibitors were peptidic, large, polar and clinically ineffective. These have been progressively improved and they have been made less peptidic. The first orally bioavailable drug-like non-peptidic BACE1-inhibitor LY28113776 was found to significantly lower Aß protein in animal models. Healthy volunteers later treated with the compound also demonstrated this decrease in Aß levels. Its safety, tolerability and efficacy were determined in a double blind, placebo controlled study in which subjects were assigned either the active drug or a placebo and participated in CSF and plasma sampling. The drug was discontinued due to its pathological effect on accumulations of autofluoroscent material within the retinal epithelium, causing them to be enlarged. Although the drug did not go on to enter the later clinical trial stages, data recorded nevertheless provides support for BACE1 as a target for BACE1 inhibitors.40
Despite support for BACE1 inhibitors, certain concerns were yet to be dealt with. In particular, it was not known whether BACE1 played other important physiological roles apart from being involved in the amyloidogenic pathway.40 In a 2006 study, it was discovered that BACE1 is involved in the myelination process of peripheral and central nerves. In BACE1-null mice, hypomyeli-nation occurred. This hypomyelination was thought to be due to the role of BACE1 in the cleavage of neureguin-1 type III, a myelination initiator. At this point, it was still unclear whether neuroglenin-1 cleavage is required for myelin sheath maturation, which would make BACE1 inhibitors potentially dangerous.41 However, it was later found that neuroglenin1 signalling is in part independent of BACE1 function. Even though BACE1's processing of the gene does produce a myelin-inducing signal, this signal is not essential for myelination stimulation.42
The first ß-secretase inhibitor to enter Phase I clinical trials was announced in 2008.2 This compound, CTS-21166, developed by CoMentis, was able to significantly lower Aß levels. It was not only potent (1.2-3.6 nm), but also selective. In cellular assays performed, CTS-21166 did not bind to 60 other enzymes.38,40 This was an achievement, as non-specific binding was one of the major issues in developing a clinically effective ß-secretase inhibitor. It also displayed desirable brain penetration, another problem associated with the development of other inhibitors (Strobel G. Keystone Drug News: CoMentis BACE inhibitor debut. http://www.alzforum.org/new/detail.asp?id=1790. Cited 2013 Sep 30). The injection of CTS-21166 into transgenic mice resulted in reduction of Aß 40 and 42 by an average of 36.5% as well as reduction in amyloid plaques in hippocampal and cortical areas by about 40%. Using this knowledge, the compound was then tested on healthy male volunteers for its tolerability and safety in 6 volunteers, with a range of doses (7.5-225 mg) given intravenously. Results were positive, showing good tolerability over the range of doses administered and slow drug clearance (Strobel G. Keystone Drug News: CoMentis BACE inhibitor debut). Significant plasma Aß inhibition continued for up to 72 hours, with recovery to normal levels by 144 hours after the inhibitor was administered.39 Later phase clinical trials have yet to be published.
γ-secretase inhibition is perhaps the most widely and frequently studied mechanism to reduce Aß. It is involved in the second step of the amyloidogenic pathway and is thus crucial in Aß generation. Another product derived from γ-secretase cleavage is the amyloid intracellular domain (AICD), which may have a role in gene expression downstream.6 γ-secretase is an intra-membrane cleaving protease, consisting of 4 components: presinilin (PS), nicastrin (NCT), presenilin enhancer (Pen2) and anterior pharynx defective (Aph1). These 4 components were thought to be essential for the activity of γ-secretase, and the loss of any of the proteins appeared to abolish the activity of γ-secretase. However, in NCT knock-out mice, it was observed that a complex of PS1, Pen2 and Aph1a was able to function as a γ-secretase inhibitor, which indicated that NCT is not required for substrate recognition in γ-secretase. Instead, it is thought to have a role in the stabilization of the enzyme. This makes it much more complex than ß-secretases.6,43 Even though NCT may not be the most important component, γ-secretase activity is PS dependent. It was the combined knowledge of mutations in PS1 and PS2 resulting in early-onset of AD and PS mutations also causing increased levels of Aß that led to the belief that PS has a direct impact in γ-secretase mediated cleavage of APP.44 In an investigation of PS-1 deficient mouse embryos, the cleavage of APP by γ-secretase was prevented, causing Aß levels to be significantly reduced, whereas cleavage by ß- and .α-secretase was unaffected.45 This thus confirmed the function of presenilin in γ-secretase.
Despite γ-secretases’ complexity, the development of γ-secretase inhibitors has been easier than the development of ß-secretase inhibitors. This is due to the hydrophobicity of its active site. Inhibitors developed were hydrophobic, which aided its permeability, and penetrating the BBB and neuronal membranes was less of a problem.6 However, its development was not without challenges. The most serious concern faced was that γ-secretase had other physiological roles in development via Notch signalling regulation. Normally, Notch is cleaved in the Golgi apparatus to produce mature Notch1 protein. Notch-1 undergoes further cleavage to release the Notch intracellular domain (NICD). NICD in turn modifies the transcription of target genes within the nucleus. In PS1 knock-out cells, it was observed that PS1 is vital in the cleavage of APP and Notch.46 Hence, the use of γ-secretases in the treatment of AD can be expected to cause gastrointestinal toxicity, immunosuppression and anaemia due to the role of Notch-1 in haematopoiesis. PS is also involved in several other processes, such as the cellular trafficking of proteins. The knock out of PS would lead to the accumulation of protein fragments within the cell due to interrupted transport between subcellular compartments. It has also been suggested that PS plays a role in homeostasis of Ca2+.47 Efforts to overcome these challenges come from the development of selective γ-secretase inhibitors that target cleavage of APP only.
Notch-sparing γ-secretase inhibitors have also been developed, and these are able to reduce APP proteolysis in the amyloidogenic pathway, thus decreasing Aß, while preventing the cleavage of Notch.47 One of the first compounds with this property was STI571 (Gleevec), an abl kinase inhibitor. STI571 was found to be able to reduce Aß production, albeit weakly, while at the same time did so without causing cleavage of Notch. These agents formed the basis of development of a number of more potent selective inhibitors, and several of such inhibitors have been reported to enter clinical trials.47,48
LY-450139 (Semagacestat), a selective γ-secretase inhibitor that is able to reduce Aß production, has been the most widely studied. LY-450139 resulted in dose-dependent decrease in Aß concentrations over a period of 6 hours post-administration, with maximum reduction of 40% relative to the baseline concentration.49 This correlated with the fact that semagacestat is absorbed rapidly from the gut, reaching its peak 1.5 hours after administration and the fact that it is mainly excreted renally. In its phase I clinical trial, manageable adverse events were reported. In the Phase II 14-week study performed in 51 mild-to-moderate cases of AD, semagacestat was shown to be safe and tolerable.50,51 However, during its Phase III trial in 2010, the drug was discontinued due to its detrimental effects on cognitive function of patients receiving it compared to those receiving the placebo. Semagacestat also appeared to be associated with increased risks of skin cancer. Such harmful effects were thought to be due to the involvement of the inhibitor with Notch cleavage and CTFß, a neurotoxic precursor of Aß (AlzForum. Drugs in clinical trials. http://www.alzforum.org/drg/drc/detail.asp?id=108. Accessed: 2 Apr 2013).2 Setbacks have also been faced in other trials, as seen in Phase III clinical trials of tarenflubril. These trials aimed to determine the efficacy, safety, and tolerability of the drug in the treatment of mild-to-moderate AD patients. However, results failed to show any benefit from tarenglubril in the cognitive function of patients. Possible reasons for this lack of success include insufficient doses and its low penetration across BBB.52
BMS-708163 (Avagacestat) is another example of a γ-secretase inhibitor. Its phase I clinical trial assessed its tolerability, safety and pharmacokinetic properties in young, healthy male volunteers. It reached its peak concentrations rapidly, in about 1.5 hours, with a plasma half-life of about 33 to 34 hours. There was a dose proportional increase in plasma concentration within the range of 0.3-800 mg of the drug. Avagacestat was able to reduce Aß by up to 88% 1 hour post-administration. Some minor side effects included nausea, dizziness and headaches, with no serious side effects or deaths reported. This phase I clinical trial thus demonstrated Avegacestat's tolerability for up to 800 mg dosage, and its suitability for progression into larger clinical studies.53 In a phase II clinical trial, 209 patients with mild-to-moderate AD were randomized to receive either the active compound or a placebo. The phase II clinical trial established that daily doses of 25 and 50 mg were indeed well-tolerated, whereas 100 and 125 mg doses were not as well tolerated and had a tendency to cause worsening of cognitive function and GI disturbances related to Notch involvement.54 These clinical trials have so far established an acceptable dose range for future studies of Avagacestat.
Taking into account the amount of research that has gone into developing secretase inhibitors, the outcome has not been desirable. The trials so far have come up empty handed, showing no therapeutic improvements in AD patients. As a result, there has been reasonable concern that anti-amyloid therapies may have no therapeutic effect in patients, which would mean decades of wasted research and possibly the targeting of the wrong compounds. Hope that anti-amyloid therapies would have a significant positive impact on AD patients is slowly dissipating. This doubt is further enhanced by the immunization of patients with full length Aß-peptides in an attempt to clear the NPs from the brain. Previously, it was unknown whether active immunization of Aß was able to benefit patients. Thus, a trial was carried out to determine its beneficial effects. In this Phase I, randomized, placebo-controlled follow up on the long term effects of Aß immunization (AN1792), data obtained indicated that immunization was indeed able to significantly reduce Aß deposition in the AD brain, in comparison to patients receiving the placebo. Disappointingly, despite the pathological improvements observed, the drug failed to produce results of cognitive improvement in these patients. On top of this, some patients even progressed to clinically severe dementia.55 What these results imply is that the removal of NPs alone may be insufficient in the disease-modifying treatment of AD. Perhaps challenges against the amyloid hypothesis are reasonable, and other factors such as tau hyperphosphorylation's contribution to AD should be re-examined in greater detail.
However, despite the disappointing lack of improvement in patients with Aß generation and deposition, there still is the possibility that these anti-amyloid strategies hold the potential to cure AD. Concluding that anti-amyloid strategies are futile at this point in time would be rash.
First of all, anti-amyloid therapies were indeed successful, as they were able to reduce NPs. Secondly, Aß deposition forming NPs was shown to be one of the earliest signs of AD, whereas cognitive decline correlated with NFT deposits.28 NPs were thought to be the earliest signs of AD, as individuals with trisomy 21 on which the gene for APP is located developed AD much earlier.56 Such observations were also made in individuals with familial AD. The initial Aß deposition is thought to set off a series of secondary downstream events, such as the accumulation of tau and inflammatory processes that cause neuronal destruction.57 Targeting NPs after a certain point would no longer be beneficial, as other processes become the main propagating factors of the disease. This indicates that perhaps the targeting of NPs much earlier on and in larger doses may be key to anti-amyloid therapies. As such, the way forward in the treatment of AD seems to be the switch from trying to cure AD to instead attempting to prevent it.
I would like to thank Prof T. Francis, PhD, Professor of Neurochemistry, Director of Brains for Dementia Research, KCL, for his guidance in this article.
The authors have no funding, financial relationships or conflicts of interest to disclose.
Conception and design the work/idea: EC. Write the manuscript: EC.
1. Povova J, Ambroz P, Bar M, Pavukova V, Sery O, Tomaskova H, et al. Epidemiological of and risk factors for Alzheimer's disease: A review. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub. 2012Jun;156(2):108–14.
2. Yiannopoulou KG, Papageorgiou SG. Current and future treatments for Alzheimer's disease. Ther Adv Neurol Disord. 2013Jan;6(1):19–33.
3. Förstl H, Kurz A. Clinical features of Alzheimer's disease. EEur Arch Psychiatry Clin Neurosci. 1999;249(6):288–90.
4. Lukiw WJ. Amyloid beta (Aß) peptide modulators and other current treatment strategies for Alzheimer's disease (AD). Expert Opin Emerg Drugs. 2012 Mar 23. [Epub ahead of print]
5. Ballard C, Corbett A. Management of neuropsychiatric symptoms in people with dementia. CNS Drugs. 2010Sep;24(9):729–39.
6. Martâinez A. Emerging drugs and targets for Alzheimer's disease; Volume 1: Beta-Amyloid, Tau protein and glucose metabolism. Cambridge: The Royal Society of Chemistry; 2010.
7. Liberati A, Altman DG, Tetzlaff J, Mulrow C, G⊘tzsche PC, Ioannidis JP, et al. The PRISMA statement for reporting systematic reviews and meta-analyses of studies that evaluate health care interventions: explanation and elaboration. PLoS Med. 2009 Jul 21;6(7):e1000100.
8. Almkvist O. Neuropsychological features of early Alzheimer's disease: pre-clinical and clinical stages. Acta Neurol Scand Suppl. 1996;165:63–71.
9. Galasko D. An integrated approach to the management of Alzheimer's disease: assessing cognition, function and behaviour. Eur J Neurol. 1998Oct;5(S4):S9–17.
10. Haupt M, Kurz A. Predictors of nursing home placement in patients with Alzheimer's disease. Int J Geriatr Psychiatry. 1993Sep;8(9):741–6.
11. Burns A. Psychiatric phenomena in dementia of the Alzheimer type. Int Psychogeriatr. 1992;4 Suppl 1:43–54.
12. Beard CM, Kokmen E, Sigler C, Smith GE, Petterson T, O'Brien PC. Cause of death in Alzheimer's disease. Ann Epidemiol. 1996May;6(3):195–200.
13. Blennow K, de Leon MJ, Zetterberg H. Alzheimer's disease. Lancet. 2006Jul 29;368(9533):387–403.
14. Holtzman DM, Bales KR, Tenkova T, Fagan AM, Parsadanian M, Sartorius LJ, et al. Apoliproprotein E isoform-dependent amyloid deposition and neuritic degeneration in a mouse model of Alzheimer's disease. Proc Natl Acad Sci U S A. 2000Mar 14;97(6):2892–7.
15. Piaceri I, Nacmias B, Sorbi S. Genetics of familial and sporadic Alzheimer's disease. Front Biosci (Elite Ed). 2013 Jan 1;5:167–77.
16. Milinois HJ, Florentin MM, Giannopoulos S. Metabolic syndrome and Alzheimer's disease: A link to a vascular hypothesis?. CNS Spectr. 2008Jul;13(7):606–13.
17. Skoog I, Gustafson D. Update on hypertension and Alzheimer's disease. Neurol Res. 2006Sep;28(6):605–11.
18. Carlsson CM. Type 2 diabetes mellitus, dyslipidemia, and Alzheimer's disease. J Alzheimers Dis. 2010;20(3):711–22.
19. Naderali EK, Ratcliffe SH, Dale MC. Obesity and Alzheimer's disease: a link between body weight and cognitive function in old age. Am J Alzheimers Dis Other Demen. 2009 Dec-2010 Jan;24(6):445–9.
20. Fratiglioni L, Paillard-Borg S, Winbald B. An active and socially integrated lifestyle in late life might protect against dementia. Lancet Neurol. 2004Jun;3(6):343–53.
21. Francis PT, Palmer AM, Snape M, Wilcock GK. The cholinergic hypothesis of Alzheimer's disease: a review of progress. J Neurol Neurosurg Psychiatry. 1999Feb;66(2):137–47.
22. Rogawski MA, Wenk GL. The neuropharmacological basis for the use of Memantine in the treatment of Alzheimer's disease. CNS Drug Rev. 2003Fall;9(3):275–308.
23. Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, et al. Correlative memory deficits, Aß elevation, and Amyloid plaques in transgenic mice. Science. 1996Oct 4;274(5284):99–102.
24. Lacor PN, Buniel MC, Furlow PW, Clemente AS, Velasco PT, Wood M, et al. Abeta oligomer-induced aberrations in synapse composition, shape, and density provide a molecular basis for loss of connectivity in Alzheimer's disease. J Neurosci. 2007Jan 24;27(4):796–807.
25. Lambert MP, Barlow AK, Chromy BA, Edwards C, Freed R, Liosatos M, et al. Diffusible, nonfibrillar ligands derived from Abeta1–42 are potent central nervous system neurotoxins. Proc Natl Acad Sci U S A. 1998May 26;95(11):6448–53.
26. Mudher A, Lovestone S. Alzheimer's disease-do tauists and baptists finally shake hands?. Trends Neurosci. 2002Jan;25(1):22–6.
27. Trojanowski JQ, Lee VMY. Rous-Whipple Award Lecture. The Alzheimer's brain: finding out what's broken tells us how to fix it. Am J Pathol. 2005Nov;167(5):1183–8.
28. Tiraboschi P, Hansen LA, Thal LJ, Corey-Bloom J. The importance of neuritic plaques and tangles to the development and evolution of AD. Neurology. 2004Jun 8;62(11):1984–9.
29. Birks J. Cholinesterase inhibitors for Alzheimer's disease. Cochrane Database Syst Rev. 2006 Jan 25;(1):CD005593.
30. Courtney C, Farrell D, Gray R, Hills R, Lynch L, Sellwood E, et al. Long-term donepezil treatment in 565 patients with Alzheimer's disease (AD2000): randomized double-blind trial. Lancet. 2004Jun 26;363(9427):2105–15.
31. Cummings J, Froelich L, Black SE, Bakchine S, Belleli G, Molinuevo JL, et al. Randomized, double-blind, parallel-group, 48-week study for efficacy and safety of a higher-dose rivastigmine patch (15 vs. 10cm2) in Alzheimer's disease. Dement Geriatr Cogn Disord. 2012;33(5):341–53.
32. Reisberg B, Doody R, Stoffler A, Schmitt F, Ferris S, Mobiue HJ, et al. Memantine in moderate-to-severe Alzheimer's disease. N Engl J Med. 2003Apr 3;348(14):1333–41.
33. Maidment ID, Fox CG, Boustani M, Rodriguez J, Brown RC, Katona CK. Efficacy of Memantine on behavioural and psychological symptoms related to dementia: a systematic meta-analysis. Ann Pharmacother. 2008Jan;42(1):32–8.
34. Zec RF, Burkett NR. Non-pharmacological and pharmacological treatment of the cognitive and behavioural symptoms of Alzheimer disease. NeuroRehabilitation. 2008;23(5):425–38.
35. Vigen CL, Mack WJ, Keefe RS, Sano M, Sultzer DL, Stroup TS, et al. Cognitive effects of atypical antipsychotic medications in patients with Alzheimer's disease: outcomes from CATIE-AD. Am J Psychiatry. 2011Aug;168(8):831–9.
36. Van Marum RJ. Current and future therapy in Alzheimer's disease. Fundam Clin Pharmacol. 2008Jun;22(3):265–74.
37. Weksler ME. The immunotherapy of Alzheimer's disease. Immun Ageing. 2004Nov 12;1(1):2.
38. Ghosh AK, Brindisi M, Tang J. Developing ß-secretase inhibitors for treatment of Alzheimer's disease. J Neurochem. 2012 Jan;120 Suppl 1:71–83.
39. Cai H, Wang Y, McCarthy D, Wen H, Borchelt DR, Price DL, et al. BACE1 is the major beta-secretase for generation of Abeta peptides by neurons. Nat Neurosci. 2001Mar;4(3):233–4.
40. May PC, Dean RA, Lowe SL, Martenyi F, Sheehan SM, Boggs LN, et al. Robust central reduction of amyloid-ß in humans with orally available, non-peptidic ß-secretase inhibitor. J Neurosci. 2011 Nov 16;31(46):16507–16.
41. Hu X, Hicks CW, He W, Wong P, Macklin WB, Trapp B, et al. Bace1 modulates myelination in the central and peripheral nervous system. Nat Neurosci. 2006Dec;9(12):1520–5.
42. Velanac V, Unterbarnscheidt T, Hinrichs W, Gummert MN, Fischer TM, Rossner MJ, et al. Bace1 processing of NRG1 type III produces a myelin-inducing signal but is not essential for the stimulation of myelination. Glia. 2012Feb;60(2):203–17.
43. Zhao G, Liu Z, Ilagan MX, Kopan R. Gamma-secretase composed of PS1/Pen2Aph1a can cleave notch and amyloid precursor protein in the absence of nicastrin. J Neurosci. 2010 Feb 3;30(5):1648–56.
44. Zhang Z, Nadeau P, Song W, Donoviel D, Yuan M, Bernstein A, et al. Presenilins are required for gamma-secretase cleavage of beta-APP and transmembrane cleavage of Notch-1. Nat Cell Biol. 2000Jul;2(7):463–5.
45. De Strooper B, Saftig P, Craessaerts K, Vanderstichele H, Guhde G, Annaert W, et al. Deficiency of presenilin-1 inhibits the normal cleavage of amyloid precursor protein. Nature. 1998Jan 22;391(6665):387–90.
46. De Strooper B, Annaert W, Cupers P, Saftig P, Craessaerts K, Mumm JS, et al. A presenilin-1-dependent gamma-secretase-like protease mediates release of Notch intracellular domain. Nature. 1999Apr 8;398(6727):518–22.
47. De Strooper B, Iwatsubo T, Wolfe MS. Presenilins and γ-secretase: structure, function, and role in Alzheimer disease. Cold Spring Harb Perspect Med. 2012Jan;2(1):a006304.
48. Netzer WJ, Dou F, Cai D, Veach D, Jean S, Li Y, et al. Gleevec inhibits beta-amyloid production but not Notch cleavage. Proc Natl Acad Sci U S A. 2003Oct 14;100(21):12444–9.
49. Siemers E, Skinner M, Dean RA, Gonzales C, Satterwhite J, Farlow M, et al. Safety, tolerability, and changes in amyloid beta concentrations after administration of gamma-secretase inhibitor in volunteers. Clin Neuropharmacol. 2005May-Jun;28(3):126–32.
50. Henley DB, May PC, Dean RA, Siemers ER. et al. Development of semagacestat (LY450139), a functional gamma-secretase inhibitor, for the treatment of Alzheimer's disease. Expert Opin Pharmacother. 2009 Jul;10(10):1657–64.
51. Fleisher AS, Raman R, Siemers ER, Becerra L, Clark CM, Dean RA, et al. Phase 2 safety trial targeting amyloid beta production with a gamma-secretase inhibitor in Alzheimer disease. Arch Neurol. 2008 Aug;65(8):1031–8.
52. Marder K. Tarenflurbil in patients with mild Alzheimer's disease. Curr Neurol Neurosci Rep. 2010Sep;10(5):336–7.
53. Tong G, Wang JS, Sverdlov O, Huang SP, Slemmon R, Croop R, Multi-center, randomized, double-blind, placebo-controlled, single-ascending dose study of the oral γ-secretase inhibitor BMS-708163 (Avagacestat): tolerability profile, pharmacokinetic parameters, and pharmacodynamic markers. Clin Ther. 2012Mar;34(3):654–67.
54. Coric v, vanDyck CH, Salloway S, Andreasen N, Brody M, Richter RW, et al. Safety and tolerability of the γ-secretase inhibitor avagescat in a phase 2 study of mild to moderate Alzheimer disease. Arch Neurol. 2012 Nov;69(11):1430–40.
55. Holmes C, Boche D, Wilkinson D, Yadegarfar G, Hopkins V, Bayer A, et al. Long-term effects of Abeta42 immunisation in Alzheimer's disease: follow-up of a randomised, placebo-controlled phase I trial. Lancet. 2008Jul 19;372(9634):216–23.
56. Lippa CF, Nee LE, Mori H, St George-Hyslop P. Abeta-42 deposition precedes other changes in PS-1 Alzheimer's disease. Lancet. 1998Oct 3;352(9134):1117–8.
57. St George-Hyslop PH, Morris JC. Will anti-amyloid therapies work for Alzheimer's disease?. Lancet. 2008Jul 19;372(9634):180–2.
Evelyn Chou, 1 King's College London, England.
About the Author: Evelyn Chou is an intercalating BSc student, after 2nd year MBBS.
Correspondence: Evelyn Chou, Address: Strand, London WC2R 2LS, United Kingdom. Email: evelynchouwy@hotmail.com
Cite as: Chou E. Alzheimer’s Disease: Current and Future Treatments. A Review. Int J Med Students. 2014 Mar-Jun;2(2):56-63.
Copyright © 2014 Evelyn Chou
International Journal of Medical Students, VOLUME 2, NUMBER 2, June 2014