Review
Fibroblast Growth Factor (FGF) Receptor Mutations: A Pathway to Understanding Multigenic
Risk in Disease?
Stuart J. Mires1
doi: http://dx.doi.org/10.5195/ijms.2013.219
Volume 1, Number 3: 123-127
Received 15 03 2013:
Accepted 21 08 2013
ABSTRACT
Fibroblast growth factor receptor (FGFR) gain-of-function mutations form the pathogenic
basis of multiple congenital pathologies. A pioneering body of work over the past
two decades has established that a unique mutation selection process within the testis
likely underlies the paternal age effect characteristics of such diseases. This mechanism,
analogous to positive selection of mutations promoting proliferation in tumorigenesis,
sparked interest in mutation profiling of testicular and other cancers. The resulting
discovery of FGFR gain-of-function mutations akin to those of congenital syndromes
has enabled a novel hypothesis to be born: that mutations represent a spectrum of
activation. As such, FGFR gain-of-function mutations could be pathogenic not solely
in defined monogenic syndromes but within myriad disease processes with ‘low activation’
conferring increased disease risk. Do such mutations contribute to multigenic risk
in multiple pathologies? This review evaluates this hypothesis, alluding to the plausible
clinical implications that ensue.
Keywords:
Fibroblast Growth Factors;
Acrocephalosyndactylia;
Craniosynostoses;
Germ-Line Mutation.
Introduction
A 31-year-old man with dwarfism; an infant with short arms, legs and a clover-leaf
skull; premature skull fusion and union of the bones of the digits in a still-born
child. Achondroplasia, thanatophoric dysplasia type II and Apert syndrome respectively,
appear to represent grossly varying pathologies at the macroscopic scale. However,
each illustrates a variant of a common pathological theme: fibroblast growth factor
receptor (FGFR) activating mutations. FGFRs are represented in multiple isoforms in
the human proteome and they have diverse functions that include cell growth and differentiation
and germ cell development. These receptors are predominant in embryological, neonatal,
and renewing tissues.1 The bodies of disease with FGFR gain-of-function (GOF) mutations as a pathogenic
agent provide a plethora of opportunity to firmly establish FGFR function.
This literature review will explore the progression of the field in characterising
the disease mechanisms conferred by such mutations. Through analysis of mutation development
at the molecular and cellular level it will unravel the complex interactions of function
in the testis and fetus, thereby establishing the concept that FGFR GOF represents
a spectrum of activation with varying contributions to pathology. Thus, it addresses
the question of whether such mutations contribute to multigenic risk in multiple pathologies.
Methodology
A literature review was performed using PubMed, MEDLINE and Embase databases, with
the free search terms ‘fibroblast growth factor receptor’, ‘gain of function’ and
‘Apert syndrome’ Additional relevant papers were retrieved from the references.
All included articles were in the English language and were relevant to FGFR gain-of-function,
identification of relation to a pathological state, or determination of plausible
mutation aetiology.
Too Much of a Good Thing?
Apert syndrome, an autosomal dominant inherited congenital malformation syndrome,
is characterised by craniosynostosis and syndactyly with a live birth rate of approximately
1 in 70,000.2 Wilkie et al., 1995 pioneered research into the condition through establishing the
molecular basis. By analysing non-recombination (alleles with the same arrangement
in affected offspring as parents) across 4 Apert families, the FGF2R locus was implicated
as the prime candidate. Amplification of FGF2R complimentary DNA (cDNA) from the patients
followed by sequencing revealed two distinct single point transversions at independent
loci in the extracellular domain, providing a putative genetic basis for the disease.3 These mutations were further illustrated to induce GOF through site-directed mutagenesis.
Protein was generated from cDNA expressing the identified Apert syndrome mutations
and complexed with FGF2 ligand. The resulting complexes were then purified and crystallised.
Both receptor mutations appeared to augment ligand-receptor interaction affinity,
providing evidence for a clinical GOF model.4
A similar body of evidence supports GOF mutations in the FGF3R transcellular domain
in achondroplasia and the FGF3R extracellular domain in thanatophoric dysplasia type
II patients.1
Are Genetic Errors Accumulated or Selected?
A well-documented characteristic of FGFR syndromes is a paternal age effect. This
stipulates that syndrome incidence increases with the age of the father at the time
of conception. Thus, a paternal inheritance was hypothesised. The presence of two
polymorphic base substitutions flanking the Apert mutation loci enabled the design
of allele-specific primers for polymerase chain reaction (PCR), comparing the haplotype
of the affected allele in patients to those of their parents. Original experimentation
identified 57/57 families showed mutations linked to the paternal allele and thus
paternal inheritance.5 This exclusivity is apparent in all further published studies to date.
A prevailing hypothesis therefore speculated that the basis of paternal inheritance
lay in the accumulation of replication-dependent mutations in spermatogonia over time,
leading to an increased frequency of mutations with paternal age and a resulting greater
likelihood of mutated spermatozoal fertilisation. This is the copy-error hypothesis.6,7
However, a fortunate experimental tool emerged when it was discovered that the Apert
mutation loci encompass the restriction site for the enzyme Microtubule organizer
protein 1 (Mbo1). Therefore, through the development of PCR primers spanning this
site, in non-mutated DNA there would be no amplification following treatment by Mbo1
due to cleavage and prevention of primer annealing whilst in mutated DNA amplification
would still occur. As such, mutation prevalence could be estimated. 6 In spermatozoa samples taken from normal men, mutation rate did not appear to vary
significantly when assessed over days, weeks or months. This implicated that mutations
were not accumulating over time. However, spermatozoa mutation level was positively
correlated with age. Further, the mutation was likely pre-meiotic since if it was
post-meiotic we would expect a reduction in mutation level with loss of spermatozoa
over time.6 Inaccuracy is present within this experimental protocol as PCR itself introduces
mutations; the efficacy of the restriction enzyme is not 100% and any mutation within
the restriction site irrespective of whether it is specific to Apert syndrome would
show as positive. Despite these limitations, this experimentation still provides strong
evidence opposing the copy-error theory.6
Further experimentation by the group utilised the same Mbo1 based technique. This
illustrated that the serine-tryptophan transversion was approximately 19-fold more
common than other mutations, thus being disproportionately high. They reasoned that
within a male heterozygous for the adjacent polymorphism to the Apert mutation locus,
if the mutation was random and accumulating it would be expected that mutations would
be equally divided between each polymorphic variant. However, the group identified
that the relative distribution was skewed. Through the use of reverse transcription
PCR (RT-PCR) analysis, expression of FGF2R RNA was confirmed in rat spermatogonial
stem cells.7 Thus, this body of evidence argues for the presence of a selection process for the
FGFR GOF mutation within the testis. Expression within spermatogonial stem cells is
a pre-requisite for selection, suggesting selection at the protein level. As such,
a novel hypothesis was born.
Spermatogonia are the basis of the stem cell capacity of the testis. It was originally
unclear whether the entire population possessed this capacity, or whether a subset
drove spermatogenesis. Through the use of genetic engineering in the mouse testis,
spermatogonia can be irreversibly labelled such that their offspring express traceable
lineage markers following exposure to tamoxifen. When pulsed with the drug, the majority
of labelled spermatozoa were lost by 2 months, due to dissociation from the seminiferous
tubule and maturation processes in the epididymis. However, a small fraction of positive
cells appeared to persist beyond 3 months, producing an average of 6.1 patches per
testis. Utilising a similar genetic labeling system, the group then went on to show
that cells isolated from these colonies, when transplanted to recipient testes, could
form de novo colonies and resulting spermatozoa, indicating a stem cell capacity.8 This experimentation presents essential evidence in supporting a selection hypothesis
within the testis. It argues for the presence of an original ‘actual stem cell’ population
generating spermatogonia which then go on to differentiate, forming spermatozoa. It
implicates that a subset of the generated spermatogonia will become ‘potential stem
cells’, deriving colonies which themselves are able to proliferate and differentiate.
Thus, if a spermatogonial stem cell population was to acquire advantageous genetic
traits, it could drive selective mechanisms through potential stem cell colony formation.
The frequency of the most common Apert mutation is 100-1,000 times higher than would
be expected from average background mutation rate. Qin et al., 2007 published pioneering
experimentation to examine the spatial distribution of mutated spermatogonia by dividing
the testes of two normal men into 200 segments and quantifying the mutation frequency
within each segment utilising PCR. This identified foci of mutation frequency 1,000-10,000
fold higher than underlying testis tissue.9 This experimentation argues for the rejection of a hypothesis that higher mutation
rates in older men are associated with Apert syndrome mutation ‘hot-spots’ (loci prone
to mutagenesis). It is highly suggestive of the selection hypothesis with mutation
foci analogous to the aforementioned potential stem cell populations.8
The ‘Selfish Testis’ and Beyond
Mouse modelling of Apert syndrome has been achieved utilising CreLox recombination
technology to generate a targeted serine-tryptophan mutation within the FGF2R. This
animal model has been intrinsic in establishing downstream effector mechanisms mediating
pathogenesis. Through the use of RNA interference knockdown of the mutated allele
in mice heterozygous for the Apert mutation, Shukla et al., 2007 illustrated that
ERK1/2 levels were normalised at the RNA and protein level compared to enhanced expression
seen in mutant animals. These members of the mitogen activated protein tyrosine kinase
family (Figure 1) were thus strongly implicated as mediators at the molecular level of GOF FGFR mutations.
Treatment of mutant mice with U0126, an inhibitor blocking activation of the extracellular
signal-regulated kinase (ERK) pathway, facilitated birth of morphologically normal
mice when injected into pregnant mothers, further supporting this pathogenic mechanism.101112 Thus, it is apparent that activating FGFR mutations appear to drive mitogenic pathways
within affected cells.
Figure 1.
Fibroblast Growth Factor Receptor Cellular Signalling
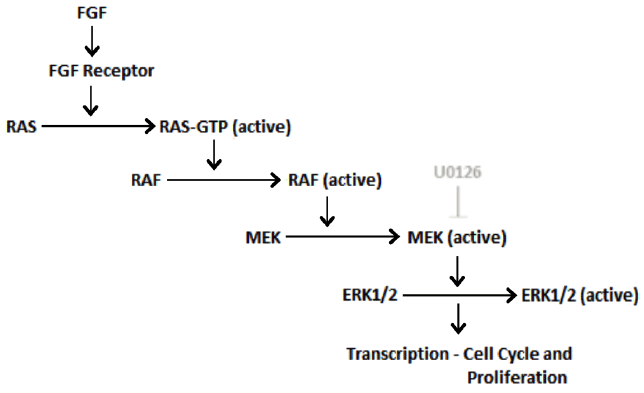
Adapted from: Seger R, Krebs EG. The MAPK signaling cascade. FASEB J. 1995;9(9):726-35;
Eswarakumar VP, Lax I, Schlessinger J. Cellular signaling by fibroblast growth factor
receptors. Cytokine Growth Factor Rev. 2005;16(2 SPEC. ISS.):139-49.
FGFR activating mutations have been identified within a range of tumour types. This
is particularly apparent in the context of endometrial carcinoma. Through sequencing
endometrial cancer cell DNA from cell lines and primary uterine tumours, it has been
identified that 30% and 10% of cancers, respectively, expressed varying FGFR activating
mutations. These included those identified in congenital syndromes such as Apert syndrome
and achondroplasia.13 Comparable data has been reported in further studies with sequencing and mass spectrometry
of endometrial cancers identifying 12.3% with FGF2R mutations, including those mediating
Apert syndrome. Similar mutations have been identified within a range of tumour types
including myeloproliferative disease, gastric, squamous cell, small cell lung and
breast cancers.14
The concepts of mutation selection within the spermatogonia of the testis in addition
to the probable involvement of mitogenic pathway activation are reminiscent of the
apparent involvement of oncogenic mutations in the tumours discussed. Therefore, it
was proposed that such mutations could drive tumorigenesis directly within the testis.
Classical seminoma germ cell tumours do not show the correct epidemiological characteristics
for paternal age effect mutations. However, spermatocytic seminomas, a rarer neoplasm,
commonly affect older men. Through sequencing FGFR mutation hot-spots within 30 samples
of tumours, two were identified as having GOF FGF3R mutations previously identified
in thanatophoric dysplasia II. Similar mitogenic pathway regulators such as Harvey
rat sarcoma viral oncogene homolog (HRAS), a factor associated with congenital Costello
syndrome characterised by skeletal and visceral morphological defects and mental retardation,
were also identified with GOF mutations in spermatocytic seminomas.15
This body of evidence proposes that GOF mutations in spermatogonia induce selective
advantage and clonal expansion, driving in situ testis tumorigenesis. However, a prominent
caveat of this experimentation, in addition to those assessing mutation profiles in
varying tumours, is the retrospective nature of the studies with no conclusive evidence
that the mutations identified are causative. FGFR GOF as such could represent acquired
mutations within the neoplasm, selected for by mitogenic advantage and acquired after
the original pathogenic event. Despite this, the work presents a viable theory of
a spectrum of pathology with a common FGFR GOF aetiology. Further, a novel, interesting
concept in the form of the ‘selfish testis’ is portrayed.16 FGFR GOF mutations appear to drive a selective advantage within the testis (spermatogonium)
itself but when transmitted to the resulting offspring they induce defects in growth
and cellular division due to imbalances in activating signals. Thus, the testis appears
‘selfish’ in selecting for an intrinsically beneficial mutation that becomes detrimental
in the fetus.
A Pathological Spectrum and Multigenic Risk
If we envisage FGFR mutations as a hypothetical spectrum with grades of activation
capacity, it is possible to classify the aforementioned pathological outcomes. ‘High
activating’ FGFR GOF mutations would be detrimental when acquired within the testis
and other tissues implicated in tumorigenesis. ‘Moderately activating’ mutations would
show the same selection mechanisms within the testis, but have negligible pathogenesis
in situ, acting as the pathogenic agent in congenital syndromes within resulting offspring.
A prominent question, therefore, becomes ‘what of ‘low activating’ mutations: Do they
exist and are they pathogenic?’
Unilateral segmented acne in a mosaic pattern has previously been described as a dermatological
hallmark of Apert syndrome.17 However, this disease has also been identified in patients without the classical
Apert syndrome presentation. Within one such patient, sequencing of the FGF2R gene
in cells isolated from the naevus demonstrated an identical serine-tryptophan GOF
mutation characteristic of Apert syndrome. Other dermatological stigmata associated
with this mutation include hypopigmentation and hypotrichosis (loss or reduction of
hair growth).17 Therefore, this discovery presents two important concepts: Firstly, despite this
patient presenting with an acquired FGFR mutation, as opposed to the congenital forms
of systemic disease previously described, the resulting findings affirm that GOF underlies
the pathological features of Apert syndrome. Secondly, it illustrates that such mutations
can alter the equilibrium of tissues constitutively regenerating throughout life,
leading to defects in structure and integrity. Thus, it is pertinent to question whether
alternative variants of the FGFR, other than those described in congenital syndromes,
could contribute to numerous disease aetiologies - particularly in tissues with renewing
capacity.
Single nucleotide polymorphisms (SNPs), as DNA sequence variations occurring at a
single base, provide an ideal tool to assess the viability of this hypothesis. Breast
tissue retains proliferative capacity throughout life, particularly in response to
hormonal change. Genome wide sequencing within breast cancer tissue specimens has
identified that variation in FGF2R loci, linked to eight SNPs, is associated with
a small but significant increased risk of developing breast cancer. Microarray data
confirmed by real-time quantitative PCR illustrated that within rare homozygous cells
for these SNPs there is amplification of FGF2R expression at the RNA level. Chromatin
immunoprecipitation ascertained that two of the SNPs resulted in an increased association
of FGF2R DNA with transcription factors such as Oct1 that are known to be associated
with proliferative capacity and tumorigenesis. Therefore, this provides a putative
mechanism by which sequence variation in the FGF2R can drive increased expression,
effectively representing a GOF of the receptor and resulting in tumorigenesis.18
The epithelial lining of the mouth and oropharynx also maintain a regenerative capacity.
A specific SNP within FGF4R has previously been associated with increased tumour cell
motility and progression within breast, head and neck, and sarcoma cancers. Recent
experimentation has focussed on the prognostic significance of SNP presence in squamous
cell carcinoma of the mouth and oropharynx. Through genotyping DNA from peripheral
blood samples of 122 patients and assessing protein expression by immunohistochemistry
in tumour cells, SNP presence was associated with lymphatic embolisation and disease-related
premature death. Thus, the SNP appears linked with poor prognosis, a phenomenon which
has previously been illustrated in a variety of tumours including lung and prostate
cancers.19
These experiments do, however, exhibit multiple caveats. Neither establishes a direct
causative link between the SNPs studied and tumour formation based on their retrospective
nature. Further, in assessment of FGF4R prognostic value, the treatment regimens of
patients and their responses to therapeutic intervention cannot be adequately controlled
between SNP positive and negative groups. In addition, the functional outcome induced
by SNP presence has as yet not been characterised, so links to GOF have not been established.
Nevertheless, the experimental data presented does illustrate that genetic variation,
even at levels as low as a single base, can alter function of the receptor complex
and contribute risk to the formation and outcome of varying pathologies. Therefore,
it is plausible that these polymorphisms represent a category of ‘low activating’
mutations, predisposing to increased disease burden within affected populations. It
remains to be established whether SNP derivation illustrates the same selection mechanisms
within the testis as seen in the GOF mutations discussed, which would be required
to justify this assertion. Further, the degree of disease association and the range
of implicated pathologies are still to be ascertained. However, this hypothesis signifies
an exciting prospect, putatively representing an underlying principle, which will
be essential in unravelling the complex issue of multigenic risk factors in disease
aetiology over the coming decades.
Conclusion
FGFR GOF mutation is implicated in numerous pathologies. By visualising GOF as a spectrum
of activity and studying genetic polymorphism we can speculate that such genetic traits
confer alteration in FGFR function and thus contribute to multigenic disease risk.
To prove this hypothesis, however, a number of questions remain to be answered. Although
a spectrum of activation provides an attractive model that conceptually relates genotype
to the clinical phenotypes produced, a direct comparison of the degree of GOF in each
pathology and predisposition is required. This includes assessment of RNA and protein
amplification in addition to the strength of ligand-receptor complex formation. Further,
selection of SNPs and other mutations related to disease risk need to be confirmed.
Finally, although the studies presented centre on tumorigenesis due to the mitogenic
activation induced by FGFR GOF mutation, relation to other common morbidities including
cardiovascular, endocrine and autoimmune disorders provides an important area of study.
Can this basic science impact on clinical outcomes? Is there a plausible bench-to-bedside
application of this evidence? Preim-plantation genetic screening for in vitro fertilisation
is performed for numerous pathologies including Huntington’s disease and cystic fibrosis
as well as genetic predispositions such as BRCA1/2 mutation. Were FGFR risk factors
to be confirmed, inclusive screening programmes could be developed. Further, as GOF
is inherited paternally, offspring are commonly heterozygous. Thus, targeted genetic
therapies such as antisense oligonucleotide-mediated knockdown or RNA interference
knockdown specifically against the mutated allele are feasible. With the advent of
these technologies, our continued understanding of FGFR contribution to multigenic
disease could spark progress in reducing clinical disease incidence and burden.
Acknowledgments
None.
Conflict of Interest Statement & Funding
The Authors have no funding, financial relationships or conflicts of interest to disclose
References
1. Webster MK, Donoghue DJ. FGFR activation in skeletal disorders: Too much of a good thing. Trends Genet. 1997;13(5):178–82.
2. Bonaventure J, El Ghouzzi V. Molecular and cellular bases of syndromic craniosynostoses. Expert Rev Mol Med. 2003;5(4):1–17.
3. Wilkie AOM, Slaney SF, Oldridge M, Poole MD, Ashworth GJ, Hockley AD, et al. Apert syndrome results from localized mutations of FGFR2 and is allelic with Crouzon
syndrome. Nat Genet. 1995;9(2):165–72.
4. Ibrahimi OA, Eliseenkova AV, Plotnikov AN, Yu K, Ornitz DM, Mohammadi M. Structural basis for fibroblast growth factor receptor 2 activation in Apert syndrome. Proc Natl Acad Sci U S A. 2001;98(13):7182–7.
5. Moloney DM, Slaney SF, Oldridge M, Wall SA, Sahlin P, Stenman G, et al. Exclusive paternal origin of new mutations in Apert syndrome. Nat Genet. 1996;13(1):48–53.
6. Goriely A, McVean GAT, Röjmyr M, Ingemarsson B, Wilkie AOM. Evidence for selective advantage of pathogenic FGFR2 mutations in the male germ line. Science. 2003;301(5633):643–6.
7. Goriely A, McVean GAT, Van Pelt AMM, O’Rourke AW, Wall SA, De Rooij DG, et al. Gain-of-function amino acid substitutions drive positive selection of FGFR2 mutations
in human spermatogonia. Proc Natl Acad Sci U S A. 2005;102(17):6051–6.
8. Nakagawa T, Nabeshima Yi, Yoshida S. Functional Identification of the Actual and Potential Stem Cell Compartments in Mouse
Spermatogenesis. Dev Cell. 2007;12(2):195–206.
9. Qin J, Calabrese P, Tiemann-Boege I, Shinde DN, Yoon SR, Gelfand D, et al. The molecular anatomy of spontaneous germline mutations in human testes. PLoS Biol. 2007;5(9):1912–22.
10. Shukla V, Coumoul X, Wang RH, Kim HS, Deng CX. RNA interference and inhibition of MEK-ERK signaling prevent abnormal skeletal phenotypes
in a mouse model of craniosynostosis. Nat Genet. 2007;39(9):1145–50.
11. Seger R, Krebs EG. The MAPK signaling cascade. FASEB J. 1995;9(9):726–35.
12. Eswarakumar VP, Lax I, Schlessinger J. Cellular signaling by fibroblast growth factor receptors. Cytokine Growth Factor Rev. 2005;16(2):139–49.
13. Pollock PM, Gartside MG, Dejeza LC, Powell MA, Mallon MA, Davies H, et al. Frequent activating FGFR2 mutations in endometrial carcinomas parallel germline mutations
associated with craniosynostosis and skeletal dysplasia syndromes. Oncogene. 2007;26(50):7158–62.
14. Dutt A, Salvesen HB, Chen TH, Ramos AH, Onofrio RC, Hatton C, et al. Drug-sensitive FGFR2 mutations in endometrial carcinoma. Proc Natl Acad Sci U S A. 2008;105(25):8713–7.
15. Goriely A, Hansen RMS, Taylor IB, Olesen IA, Jacobsen GK, McGowan SJ, et al. Activating mutations in FGFR3 and HRAS reveal a shared genetic origin for congenital
disorders and testicular tumors. Nat Genet. 2009;41(11):1247–52.
16. Wilkie AOM. Bad bones, absent smell, selfish testes: The pleiotropic consequences of human FGF
receptor mutations. Cytokine Growth Factor Rev. 2005;16(2):187–203.
17. Melnik BC, Vakilzadeh F, Aslanidis C, Schmitz G. Unilateral segmental acneiform naevus: A model disorder towards understanding fibroblast
growth factor receptor 2 function in acne? Br J Dermatol. 2008;158(6):1397–9.
18. Meyer KB, Maia AT, O’Reilly M, Teschendorff AE, Chin SF, Caldas C, et al. Allele-specific up-regulation of FGFR2 increases susceptibility to breast cancer. PLoS Biol. 2008;6(5):1098–103.
19. Dutra RL, de Carvalho MB, Santos Md, Mercante AMdC, Gazito D, de Cicco R, et al. FGFR4 Profile as a Prognostic Marker in Squamous Cell Carcinoma of the Mouth and Oropharynx. PLoS ONE. 2012;7(11):e50747.
Stuart J. Mires, 1 University of Oxford, Oxford, England, UK
About the Author: Stuart J. Mires is a 5th year medical student at the University of Oxford, Oxford,
England, UK.
Correspondence Stuart J. Mires, Address: Osler House, John Radcliffe Hospital, Headley Way, Headington,
Oxford, UK. OX3 9DU. Email: stuart.mires@sjc.ox.ac.uk
Cite as: Mires SJ. Fibroblast Growth Factor (FGF) Receptor Mutations: A Pathway to Understanding Multigenic Risk in Disease? Int J Med Students. 2013;1(3):123-7.
Copyright © 2013 Stuart J. Mires
International Journal of Medical Students, VOLUME 1, NUMBER 3, December 2013